
TBK1 通过增强雌激素受体α的转录活性参与乳腺癌发生的分子机制
2014-11-29 08:31郝钦芳邹德勇张丽萍张小丽葛晓幸杨晓莉
生物技术通讯 2014年6期
郝钦芳,邹德勇,张丽萍,张小丽,葛晓幸,杨晓莉
武警总医院 检验科,北京 100039
IKK 相关激酶是最近发现的功能和IKK(inhibitor of NF-κB kinase)相近的激酶,包括TANK 结合激酶1(TANK-binding kinase 1,TBK1)和IKKi(又称为IKKε),均为丝氨酸激酶[1]。TBK1包含729个氨基酸残基,其N 端1~290 位有一个激酶结构域,在激酶结构域之后的299~383 位之间存在一个泛素样结构域(ubiquitin-like domain,ULD),此外在603~650位及679~712 位包含2 个螺旋-环-螺旋(helix-loophelix,HLH)结构域。激酶结构域是TBK1 发挥其丝氨酸磷酸化的重要结构域,而ULD 则与其激酶活性、与底物结合相关[2]。TBK1 几乎在所有组织中稳定表达,Northern 印迹分析表明,TBK1 在胃、小肠、肺、皮肤、脑、心脏、肾脏、肝脏、脾脏、胸腺等器官中广泛表达[3]。
雌激素受体(estrogen receptors,ER)包括ERα和ERβ,它们都属于核受体超家族[4]。ER 能够激活含有雌激素应答元件(estrogen responsive element,ERE)基因的转录,从而引起细胞的恶性转化[5]。ERα在乳腺癌的发生发展过程中起关键作用。研究表明,ERα的磷酸化可以影响其转录激活活性[6]。ERα磷酸化可影响ERα与DNA 和配体的结合能力以及ERα的稳定性,进而影响其转录激活功能活性。ERα的Tyr537的磷酸化可调节ERα的二聚体化和DNA 结合能力,Ser118磷酸化可调节ERα的转录激活功能。用定向性位点突变试验检测ERα磷酸化后的活性,发现Ser118突变为Ala 后ERα的活性降低约25%,但与Ser104、Ser106同时突变时ERα活性可降低50%[7]。总之,这些研究表明ERα的磷酸化影响其转录激活活性。
因为TBK1 具有激酶活性,而ERα又受磷酸化调控,我们推测TBK1 可能参与调控ERα。在本研究中,我们在细胞体系中过表达TBK1,发现其可以增强ERα的转录活性,而这与TBK1 在先天免疫途径中的功能无关,而且TBK1 可以通过磷酸化ERα Ser305位调控其下游相关基因的表达。
1 材料与方法
1.1 材料
HEK293T、MCF-7 细胞购自中国科学院上海细胞库;大肠杆菌DH5α感受态菌株购自北京天根生化科技有限公司;Flag-ERα及其突变体、HA-ERα以及ERE-Luc 由军事医学科学院生物工程研究所叶棋浓研究员惠赠;Flag-TBK1 和HA-TBK1 及其突变体由本室构建;抗Tubulin、抗Flag 购自Sigma 公司;HRP标记的羊抗小鼠二抗及羊抗兔二抗购自中杉金桥生物科技公司。
1.2 转染质粒的制备
将新鲜的质粒转化菌液接种至5~8 mL 含有抗生素的LB 培养基中,37℃、200 r/min 过夜培养16~18 h,用天根公司的普通小提质粒试剂盒提取质粒,用分光光度计测定质粒的D260nm和D280nm值,计算质粒浓度,并依据D260nm/D280nm比值判断所提质粒的纯度。
1.3 细胞培养、传代和转染
细胞均用含10%胎牛血清的DMEM 培养基,在含5% CO2的恒温孵育箱中培养,采用胰酶消化法传代。传代时先吸去培养基,加入PBS清洗细胞;加入1 mL 胰酶消化液,轻轻摇动培养皿,使胰酶消化液覆盖细胞表面,室温放置3~5 min(根据不同细胞);之后加入3 mL含10%胎牛血清的DMEM培养基,吹打细胞并形成细胞悬液;将细胞加入15 mL 离心管中,1000 r/min 离心至管底,用新鲜DMEM 培养基重悬细胞,并加入新的细胞培养皿中继续培养。冻存细胞株时,先用胰蛋白酶消化细胞,同上将细胞离心至15 mL 离心管底部,用含10%二甲基亚砜(DMSO)、20%胎牛血清、70% DMEM 培养基的冻存液重悬细胞,之后将含有细胞的冻存液分装至细胞冻存管中,做好标记,先置于-70℃过夜,次日取出并投入液氮罐中保存。
利用转染试剂进行细胞转染。6 cm 培养皿中细胞生长至60%~80%汇合度时进行转染。转染前1 h 更换2 mL 新鲜的DMEM 培养基,将1 μg 质粒和2 μL 脂质体分别用生理盐水稀释,混匀后静置5 min,将含有脂质体的稀释液逐滴加至含有质粒的稀释液中,混合后室温放置15~20 min,使质粒与脂质体形成复合物,之后加入细胞中,轻轻晃动培养皿使之混匀,继续培养4~6 h 后更换新鲜培养基,24~48 h后收集细胞。
1.4 萤光素酶报告基因检测
用12孔细胞培养板培养细胞,转染24 h后将待测细胞中的培养基吸去,每孔加入1 mL PBS 漂洗细胞,之后每孔加入100 μL 1×裂解缓冲液(Promega公司),将12孔板置于摇床中,室温裂解10 min后12 000 r/min 离心1 min,将上清转移至新管中;将40 μL 细胞裂解液加入新试管中,之后在管中加入50 μL 萤光素酶检测试剂(LAR),混匀后用萤光光度检测仪检测发光值(如果所测得数值不在量程之内,则须调节萤光光度计的灵敏度),然后加入50 μL 反应终止液,并测定海肾萤光素酶的发光值作为内参,两者的比值即为萤光素酶的活性。
1.5 免疫印迹实验
将收集的细胞在4℃预冷的离心机中2000 r/min 离心5 min,弃尽上清;用1 mL 预冷的1×PBS 洗涤细胞2 次,吸尽PBS;加入凝胶上样缓冲液,100℃沸水浴5 min,离心后进行SDS-PAGE,电压为80~120 V,具体操作参照《分子克隆实验指南》(第3版)。
将PVDF 膜在甲醇中浸泡30 s,之后与2 张2.5 mm厚的滤纸在半干转移缓冲液中浸泡;将电泳结束后的SDS-PAGE 胶浸入半干转移缓冲液中,自下而上按照滤纸-胶-膜-滤纸的顺序放置在半干转移仪(Bio-Rad 公司)的2 块电极之间,尽量赶尽胶和PVDF 膜之间的气泡;设置转移电压18 V,转移时间1.5 h;转移结束后将PVDF 膜放入含5%脱脂奶粉的1×TBST 缓冲液中封闭1 h,之后用含1×TBST 或含5%脱脂奶粉的1×TBST,或者直接用1×PBST 配制的抗体(一抗)室温孵育PVDF膜1 h或过夜孵育,结束后用TBST 洗3次,每次5~10 min;一抗孵育结束后,按需加入辣根过氧化物酶(HRP)标记的抗体(二抗),室温孵育1 h,用同上方法洗膜;每种抗体孵育结束后回收抗体,进行化学发光,用X线胶片曝光。
1.6 细胞总RNA的提取及RT-PCR
由于RNase极易造成污染,故首先应当用DEPC浸泡处理所有容器以去除RNase。各种枪头和EP管均使用无RNase产品(Axygen公司)。
用10 cm 细胞培养皿培养细胞,待细胞铺满后提取总RNA。首先吸去培养基,用PBS清洗细胞,加入1 mL TRIzol 试剂(Invitrogen 公司)处理细胞,将所有液体转移至新的EP 管中,14 000 r/min 离心10 min,将上清转移至新的EP 管中合并,加入0.2 mL 氯仿,剧烈振荡15 s 后静置3 min,在预冷的离心机中离心15 min,小心吸取上层水相至新的EP管中,在收集的液体中加入0.5 mL 异丙醇,-20℃低温冰箱中放置10 min(可在冰箱中长期保存)使RNA析出,离心10 min,小心倒去上清,用DEPC 水配制的75%乙醇洗涤,14 000 r/min离心5 min后弃尽上清,在无RNase 的通风橱中干燥,但不能吹得太干,将白色沉淀物在100 μL DEPC 处理过的水中溶解,用分光光度计测量D260nm值,并计算RNA 含量,之后进行琼脂糖凝胶电泳,观察5S、18S、28S 核糖体rRNA条带,用于判断提取的RNA的质量。
之后进行反转录反应,反应体系包括总RNA 1 μg、oligo(dT)(50 μmol/L)1 μL、RNase 抑制剂1 μL、反转录缓冲液5 μL、dNTP(2 mmol/L)5 μL、反转录酶(M-MLV)0.5 μL,加DEPC 水至25 μL。在PCR 仪中进行RT-PCR 反应,反应程序设置为42℃1 h、95℃5 min。反转录得到的cDNA 可用于后续特异性PCR反应。
PCR反应体系包括cDNA模板5 μL,10×PCR 反应缓冲液5 μL,10 mmol/L dNTP 混合物1 μL,上、下游引物各1 μL,普通高效Taq酶(TaKaRa 公司)1 μL,加入去离子水补至50 μL。在PCR 仪上设置反应条件为:95℃5 min;95℃30 s,55℃30 s,72℃1 kb/min,循环次数根据不同基因的丰度调整,一般为14~22个循环;最后72℃延伸5 min。利用β-actin引物扩增β-actin,作为内参。qPCR引物见表1。
1.7 统计学处理
2 结果
2.1 TBK1可以增强ERα的转录活性
为了研究TBK1 是否能调控ERα的转录活性,我们在HEK293T 细胞中瞬时转染HA-ERα/Flag-TBK1 和ERE-Luc 报告基因(ERα转录结合元件,此报告基因的激活表示ERα入核启动转录)及pRL 质粒,选用HA-Vector(vector)/Flag-Vector(Ctrl)作为对照,检测TBK1 对ERα转录活性的影响。实验结果表明,与阳性对照组Flag-Vector(Ctrl)和HA-ERα相比,共转Flag-TBK1 和HA-ERα能够更为显著地增强ERE 的转录活性;而作为阴性对照,共转HAVector(vector)和Flag-Vector(Ctrl)或共转HA-Vector(vector)和Flag-TBK1 时则几乎不能增强ERELuc报告基因的转录活性(图1A)。为了检测这种转录活性增强作用是否具有特异性,是否与雌激素的刺激有关,我们又选择了能表达内源ERα的乳腺癌细胞系MCF-7,并在体系中加入雌激素(E2)刺激,瞬时转染Flag-TBK1,选用Flag-Vector(vector)作为对照。实验结果显示,在MCF-7 细胞中无论是否加入E2,TBK1都可以增强ERα的转录活性,而加入E2后TBK1的激活作用显著增强(图1B),说明TBK1对于ERα转录活性的增强具有特异性,既是雌激素依赖的,又是雌激素非依赖的。
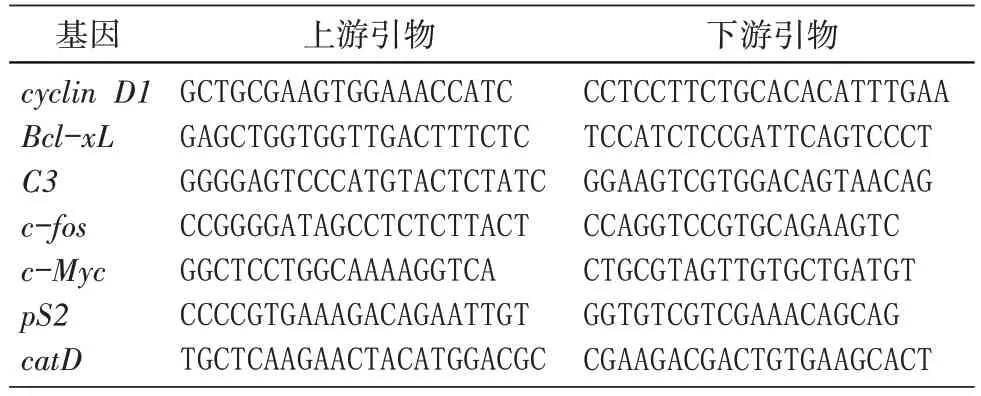
表1 引物及序列
2.2 TBK1 增强ERα转录活性的功能不依赖于其激活先天免疫途径的功能
为了研究TBK1 增强ERα转录活性的功能是否与其激活先天免疫途径的功能相关,我们首先将Flag-TBK1 或Flag-TBK1(L352A,I353A)突变体(此突变体为实验室保存,依照文献报道,会丧失激活先天免疫途径的功能)分别与IFN-β-Luc、IRF3-Luc、NF-κB-Luc 报告基因及pRL 质粒共转染HEK293T细胞,同时转染Flag-Vector(vector)作为阴性对照,24 h后收集细胞检测荧光素酶活性,并通过Western印迹检测相关蛋白的表达。结果显示,野生型TBK1能明显激活IFN-β、IRF3 和NF-κB,而Flag-Vector(vector)阴性对照和Flag-TBK1(L352A,I353A)突变体几乎不能激活IFN-β和NF-κB 信号传导通路(图2),说明TBK1 的这2 个位点突变后使其丧失了激活先天免疫途径的功能。
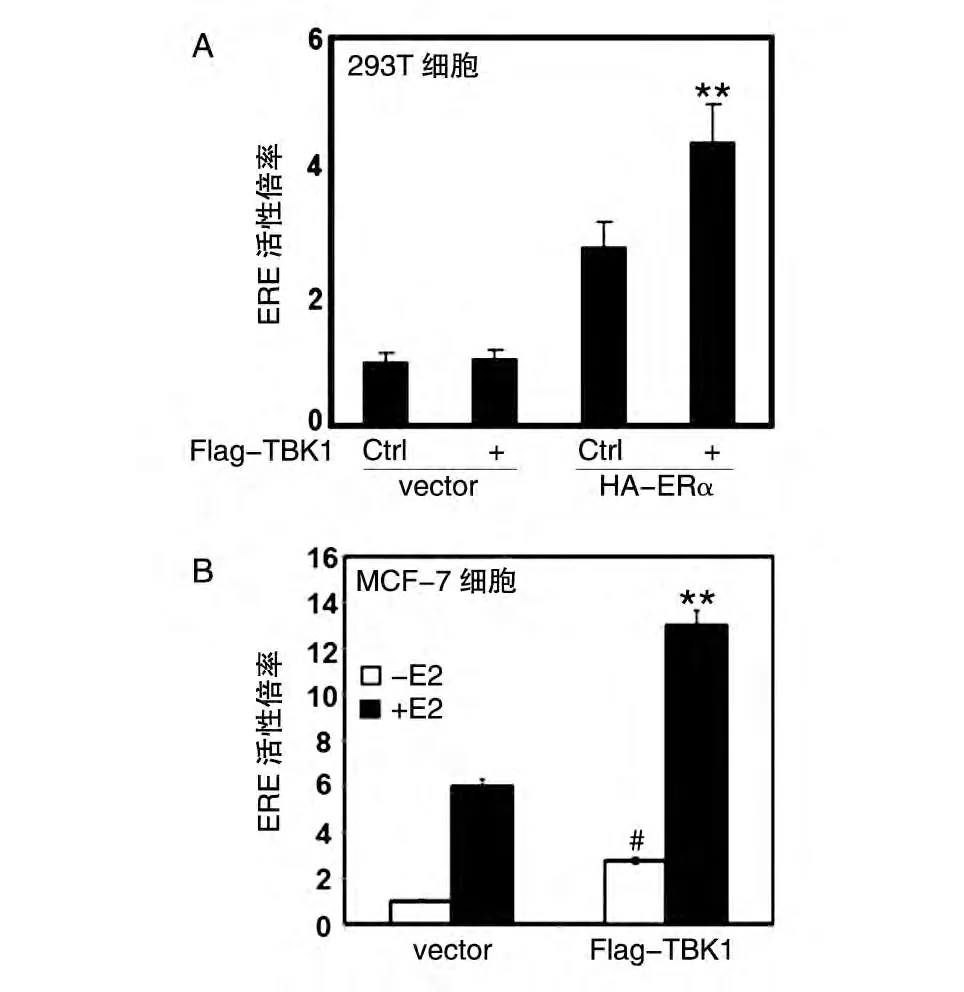
图1 TBK1增强ERα的转录活性
接着,我们在MCF-7 细胞中瞬时转染Flag-TBK1/Flag-TBK1(L352A,I353A)和ERE-Luc 报告基因及pRL质粒,检测TBK1及其先天免疫途径功能缺失突变体对ERα转录活性的影响,并用Western印迹检测各蛋白的表达水平。结果显示,TBK1(L352A,I353A)与野生型TBK1 一样,都可以明显增强ERα的转录活性(图3),说明TBK1 对ERα转录活性的激活不依赖于其激活先天免疫途径的功能。加入TBK1 的抑制剂BX795 后,TBK1 对ERα的激活作用受到明显抑制证明了TBK1 对ERα的转录激活作用。为了探讨TBK1 的激酶活性对其激活ERα转录活性的功能是否有影响,我们转入Flag-TBK1(K38A)质粒(激酶活性缺失突变体),检测其对ERα的转录活性是否具有激活作用,发现其对报告基因的激活作用降至野生组的1/2 水平(图3),说明TBK1 的激酶活性对其激活ERα转录活性的功能具有重要作用。
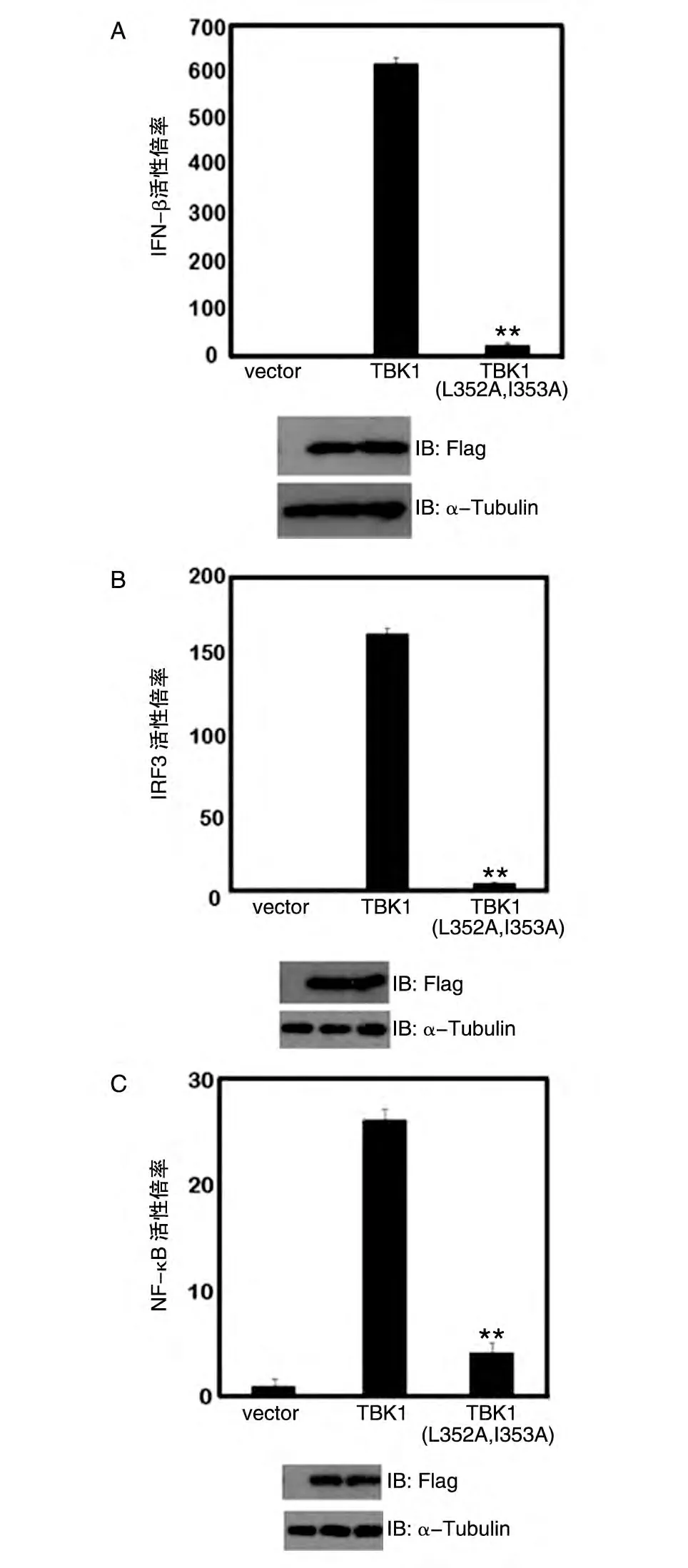
图2 TBK1(L352A,I353A)不能激活先天免疫途径
2.3 TBK1通过磷酸化修饰ERα的S305位增强其下游基因的表达
为了进一步证明TBK1 对ERα转录活性的激活作用,寻找可能的作用机制,我们将TBK1 与ERα/ERα(S305A)突变体(磷酸化位点突变)共转HEK293T 细胞,用RT-PCR 实验检测ERα下游基因的表达。结果表明,在野生型ERα存在时,TBK1 能够明显增强ERα下游基因的表达;在ERα(S305A)突变体存在时,TBK1却不能增强ERα下游基因的表达(图4)。野生型ERα下游基因的表达比磷酸化位点突变的ERα(S305A)下游基因的表达水平高(野生型ERα组的cyclin D1表达水平是突变体组的近3倍,Bcl-xL的表达水平是突变体组的近2倍,C3的表达水平是突变体组的近5 倍,c-fos的表达水平是突变体组的3 倍多,c-Myc的表达水平是突变体组的4倍,pS2的表达水平是突变体组的3 倍多,catD的表达水平是突变体组的近4 倍),不但证明了TBK1 对ERα转录活性的增强作用,而且证明这种作用是依赖于TBK1对其S305位的磷酸化修饰实现的。
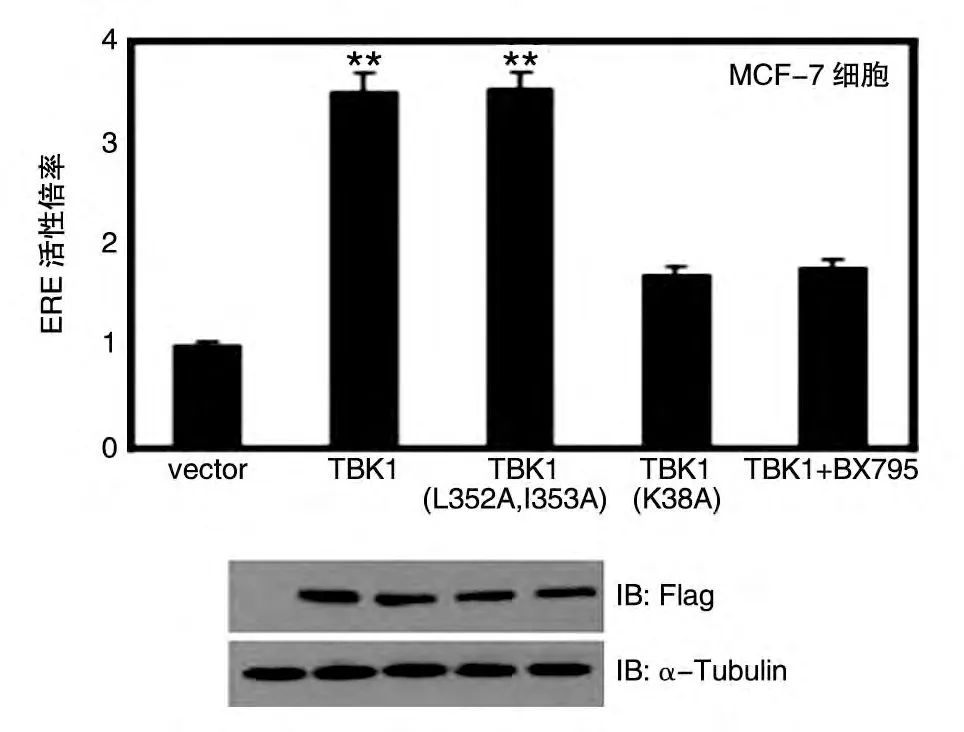
图3 TBK1(L352A,I353A)能够激活ERα的转录活性
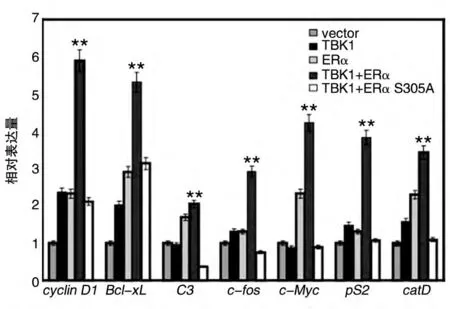
图4 TBK1通过磷酸化修饰ERα S305位增强其下游基因的表达
3 讨论
乳腺癌是女性最常见的恶性肿瘤之一,雌激素在乳腺癌的发生发展中有极其重要的作用。转录因子ERα通过与雌激素结合,调控含ERE 基因的表达,因此,ERα在乳腺癌的发生发展过程中起重要作用。本研究的主要发现包括:TBK1可以增强ERα的转录活性;将TBK1 进行L352A、I353A 位点突变后,其丧失激活先天免疫途径功能,但仍然能够增强ERα的转录活性,说明此作用不依赖于其在先天免疫途径中的功能;TBK1 可以通过磷酸化ERα的S305 位激活ERα,使其入核启动下游基因的转录。这些发现进一步阐明了乳腺癌发生的机制,对乳腺癌的治疗和靶向药物开发具有一定的指导意义。
TBK1中有一个重要的泛素样结构域,研究发现ULD 对于TBK1 的磷酸化活性以及其和下游磷酸化底物的结合有着重要的作用[1]。当TBK1 的ULD 被突变之后,TBK1 就失去了与下游激酶底物IRF3 及IκBα结合的能力,从而失去激活下游Ⅰ型干扰素分泌及NF-κB 信号通路的功能。我们将ULD 中重要的352 位Leu 和353 位Ile 都突变为Ala,发现突变体组相比野生型TBK1组几乎不能激活IFNβ及NF-κB信号传导通路,但依然能够激活ERα的转录活性,这就表明TBK1 参与ERα信号通路并不依赖于其对NF-κB 通路及IFN-β通路的激活,这些研究初步阐明了先天性免疫信号通路和乳腺癌发生之间的关系。
[1]Hacker H,Karin M.Regulation and function of IKK and IKK-related kinases[J].Sci STKE,2006,2006(357):re13.
[2]Peters R T,Maniatis T.A new family of IKK-related kinases may function as I kappa B kinase kinases[J].Biochim Biophys Acta,2001,471:M57-62.
[3]Pomerantz J L,Baltimore D.NF-kappaB activation by a signaling complex containing TRAF2,TANK and TBK1,a novel IKKrelated kinase[J].EMBO J,1999,18:6694-6704.
[4]Osbome C K,Elledge R M,Fuqua S A W,et al.Estrogen receptors in breast cancer therapy[J].Sci Am,1996,9:32-42.
[5]Turner R T,Riggs B L,Spelsberg T C,et al.Skeletal effects of estrogens[J].Endocr Rev,1994,15:275-300.
[6]Nilsson S,Gustafsson J A.Estrogen receptor transcription and transactivation:basic aspects of estrogen action[J].Breast Cancer Res,2000,2:360-366.
[7]Yamashita H,Nishio M,Toyama T,et al.Low phosphorylation of estrogen receptor alpha(ERalpha) serine 118 and high phosphorylation of ERalpha serine 167 improve survival in ER-positive breast cancer[J].Endocr Relat Cancer,2008,15(3):755-763.
猜你喜欢
河北北方学院学报(自然科学版)(2022年11期)2022-02-03
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国科技纵横(2021年24期)2021-03-02
中成药(2018年6期)2018-07-11
生命科学研究(2018年1期)2018-05-29
安徽医科大学学报(2016年12期)2017-01-15
中国医药生物技术(2015年4期)2015-12-26
天津医科大学学报(2015年2期)2015-12-22
山东医药(2015年40期)2015-02-28
