
肿瘤微阵列数据统计分析概述
2014-11-29 04:16张杨张威曹文君李运明陈长生
生物技术通讯 2014年6期
张杨,张威,曹文君,2,李运明,3,陈长生
1.第四军医大学 军事预防医学院卫生统计学教研室,陕西 西安 710032;2.长治医学院 基础部,山西 长治 046000;3.成都军区总医院 神经外科,四川 成都 610083
随着生物技术和统计方法的发展和改进,微阵列技术可以在一次试验中对整个基因组进行分析,已广泛应用于分析大规模的mRNA 表达数据。目前有4 类生物芯片平台被广泛应用,即Affymetrix GeneChip 芯片、寡核苷酸探针芯片、cDNA 芯片和商业化探针芯片。在肿瘤研究领域,微阵列技术对肿瘤的诊断和分型、治疗和预后,以及探讨肿瘤发生的分子机制和发展都有非常重要的作用。基因表达谱数据分析,对肿瘤患者的个性化治疗和肿瘤的分子分型发挥着越来越重要的作用。
由于微阵列技术所得到的基因表达谱数据具有高维(成千上万个基因)和样本量小的特点,如何挖掘和解释其中所蕴含的海量基因信息,深层次研究基因功能,选择适当的统计学方法对于芯片数据的处理至关重要。微阵列基因表达谱数据信息的提取及其统计分析方法的研究,已成为生物与医学统计学领域中富有挑战性的重要课题。
1 肿瘤研究中微阵列技术的流程
微阵列试验的基本过程如图1 所示。基因表达谱通过计算机软件扫描图像提取得到的原始数据首先通过标准化方法过滤掉那些低质量的探针数据,即在所有样本中都表现出较低的信号强度或变化幅度,对感兴趣的表型或条件不可能有贡献[1]。芯片数据只有通过标准化处理后,才能进行下游的统计分析,如筛选差异表达的基因或肿瘤分型。微阵列技术通常被作为筛选工具,生物验证和解释应对选定的基因做进一步研究。
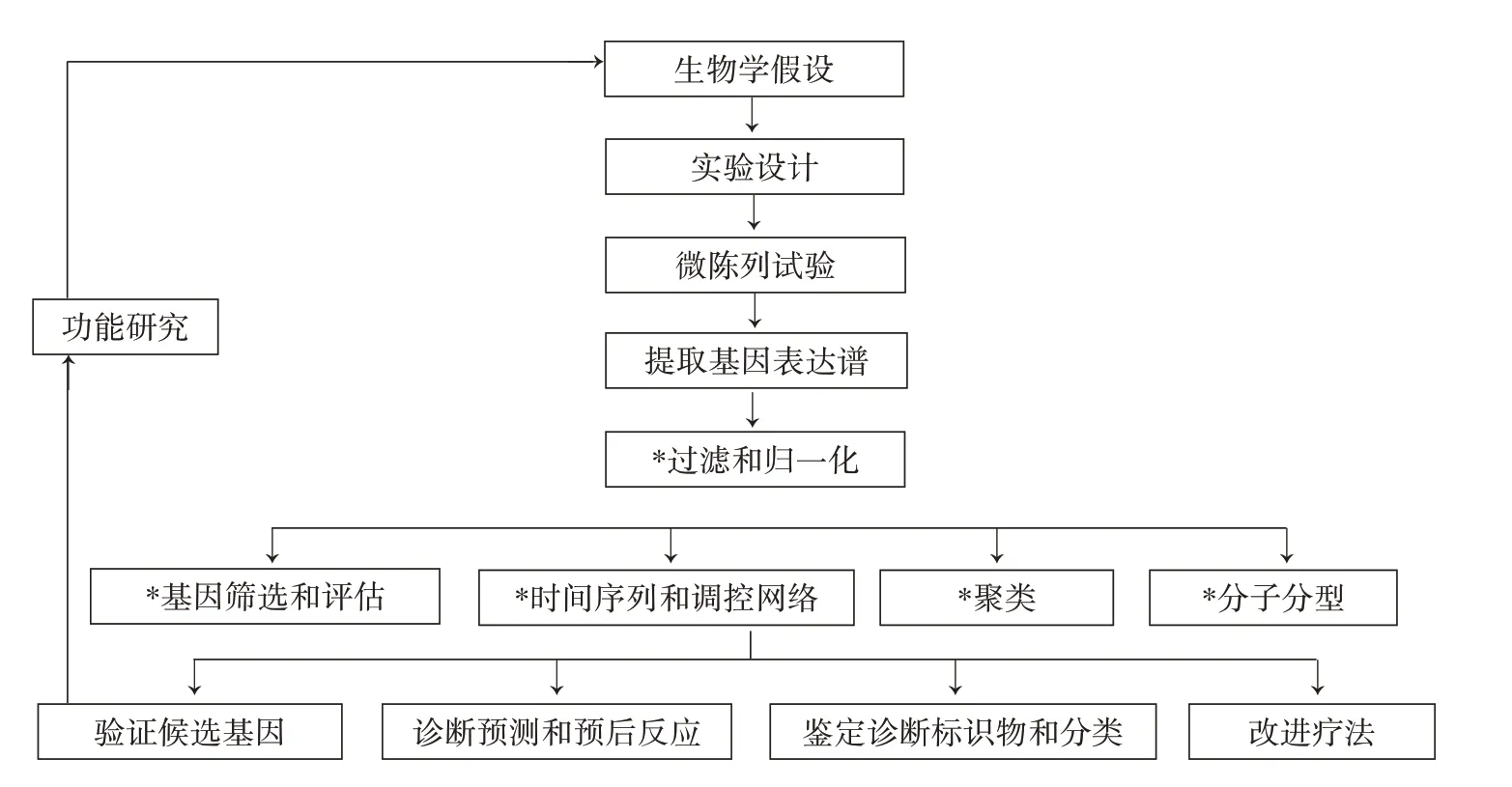
图1 微阵列试验流程图,其中标星号的流程为统计分析部分
2 cRNA微阵列数据的标准化和预处理
微阵列数据的质量对下游的统计分析至关重要,包括RNA 的质量、探针标记、杂交条件、洗板,以及在扫描当中的信号强度和背景干扰。尤其在低丰度表达RNA 分子的研究中容易受背景系统的影响而导致系统偏差。这些偏移可导致基因表达数据研究的错误结论,即假阳性和假阴性的预测。但这些变异和微阵列数据的系统性偏差可通过科学的重复和归一化进行控制。
在实验过程中,当我们从实验组和对照组样本中获得基因表达谱后,可以通过计算每个基因在Cy3和Cy5 通道的荧光染料强度的平均对数比值而得到该基因的表达强度[1]。通过比较在不同处理组中该基因表达强度的差异,对其进行进一步的研究。在得到该表达强度的数据后,首先要进行的是将低重复性的探针数据过滤去除。该过程可以通过控制编译系数(小于特定的阈值)和表达密度(大于特定表达水平)进行。经过预处理后,扫描图像的整体亮度和试验变化所造成的系统性偏差可以得到有效控制。例如,模块和染料的影响。此步骤对微阵列数据的多重比较和下游的统计分析是必不可少的。这一过程统称为归一化。
归一化的方法很多,其中应用最广泛的是全局归一化(global normallization)。该方法的目的是将所有芯片探针均归一化为具有相同的中位表达强度,这一方法可以很好地矫正模块数据。然而,大量的统计学研究证明模块数据的影响是存在的。针对这一影响,Dudoit 等经过不断研究,发展了对每个模块强度数据归一化方法,称为“LOESS归一化”[1]。基本生物学假设是,在一个模块中上调基因和下调基因的表达量是基本一致的。由于这一归一化方法的假设条件,因此不适用于定制芯片和经处理的特定细胞系表达数据的研究。在该方法的基础上,Tseng等[2]使用“不变基因集”作为看家基因的代理,并仅基于该不变基因集的强度来估计模块强度。Fan 等[3]在没有以上方法的生物学假设条件下,利用阵列内重复,提出切片内半线性模型(semilinear in-slide)归一化方法。该方法基于芯片内约100 次的重复探针,以避免序列特异性和噪音干扰。其基本依据是,在同一模块中重复探针的基因表达差异基本反映了除随机噪音之外的系统误差,而这些系统误差可以通过探针配对加以去除。使用切片内半线性模型,通过选取神经瘤细胞转移抑制因子靶向基因,以实时反转录PCR 反应被加以验证,而普通的归一化方法则容易造成一些基因的缺失。随后,Fan 等[4]通过芯片内重复探针和联合其他芯片的信息,显著扩大了这一方法的适用范围。除了以上归一化方法外,其他有用的归一化方法还包括双向半线性模型[5]和稳健(robust)归一化等[6]。
3 芯片内重复
芯片内重复不仅对归一化的作用很大,而且对于验证数据归一化是否正确也非常有用。其基本的思想是,芯片内重复之间的差异是系统偏差除去后的纯粹的随机噪声。当估计每个单独基因的噪声水平并且芯片通过归一化,那么总的标准化方差大概服从卡方分布。这对检测数据是否已归一化提供了一个简单而有用的诊断测试方法。这一检验统计量也可作为一个给定阵列选择归一化方法的标准——最小的检验统计量(最一致的重复)是最优的选择。可以通过过滤方法和经验贝叶斯的方法估计相应的方差的差异[7]。芯片内重复也被用于改进基因集方差估计的精度,从而提高推断方法的设计来识别差异表达基因[8]。
4 筛选差异表达基因
微阵列实验的主要目的就是筛选实验组和对照组或者更复杂的比较组间的差异表达基因[9]。选择恰当的统计学方法在此过程中至关重要。首先,是选择恰当的检验统计量,通常是经过修正的检验统计量,如在微阵列显著性分析中,修正过的单样本和两样本t检验、方差分析或经典贝叶斯方法[10]。由于微阵列数据的一个显著特点是一般只有一小部分的基因是差异表达的,因此,根据不同芯片本身的特点选择合适的检验统计量,以提高芯片数据处理的灵敏度和特异度。
在选择好检验统计量之后,下一个步骤是计算检验统计量并由此得出显著性P值。芯片数据常常要同时处理成千上万个基因,相应的P值的计算也较为复杂。例如,当样本量较大,且服从正态分布时,可采用student't 分布以计算P值;而在小样本非正态分布时,permutation 或bootsrapping 方法是较为合适的选择。
判断出P值之后,接下来将是筛选差异表达的基因。由于微阵列试验同时检验成千上万个基因,很可能出现较高的阳性结果错误发现率。因此,控制错误发现率对结果的生物学解释至关重要[11-12]。目前已发表了很多关于控制错误发现率方法的论文。Storey等[11]和Dudoit等[12]对关于基因集中控制错误发现率的方法进行了综述。这些方法要求非常精确地P值,通常在10-6数量级。此外,有些方法可以通过判断检验统计量是否超过某一界值或相应的P值(小于阀值)来筛选差异表达基因,从而控制错误发现率[2,10]。例如,假设在15 000 个基因中有100 个基因相应的P值小于0.001,则期望的被错误发现的基因数不超过0.001×15 000=15,那么我们可以估计错误发现率为15/100=15%[2]。
5 微阵列技术在肿瘤分类和聚类中的应用
微阵列技术肿瘤临床研究的一个重要应用是发现肿瘤生物学标志物和对肿瘤进行病理分类。Inamura 等[13]在其研究中对这一领域进行了较为详细的阐述。他们在肺癌和正常肺组织标本的研究中,通过分层聚类和非负矩阵因子化的方法将肺鳞状细胞癌分为2个不同的亚型,2个亚型具有完全不同的分子特征和临床结局。
分类也被称为有监督学习。许多统计分析方法被应用于聚类和分类,这些方法包括决策树分类方法、线性鉴别分析、支持矢量机法,以及神经网络特征分析[14]。使用归一化芯片数据作为输入向量,可以建立分类规则。Svrakic 等[15]对基因集中所应用的聚类方法做了全面综述。
通常我们在对肿瘤的分类研究中,希望在筛选基因或肿瘤标志物的过程中得到具有较高判别效能和低误判率的差异表达基因。这不仅提高了基因在肿瘤中功能的理解,而且降低了错误分类的几率。基因表达谱收缩因子方法在肿瘤分类研究中被作为一种重要的分类方法[16]。统计变量选择法,如误判的出发散度,也可被应用于筛选重要的差异表达基因和肿瘤标志物[17]。
聚类也称为无监督学习方法,常常用于对基因表达谱中具有相似表达特征的基因进行归类[15]。这有利于我们发现共表达或表达特性相似的基因群。聚类算法的一个重要步骤是如何在输入空间中定义合适矩阵指标,如分层和K最近邻分类法[15-16]。这些输入向量既可以是不相关的基因集中具有相似表达特性的基因,也可以是不同样本的同一表达基因。在聚类过程中,常用的计算指标包括欧氏距离和Pearson 相关系数。相似表达基因通常通过系统树图或彩色编码表示。
6 时间序列和调控网络
为了监测基因的队列表达模式,基因随着疾病的进展时间,或在不同治疗中的表达情况,我们可以在不同的时间点取样获得基因的表达数据。统计学上一个重要的问题是,在某个特定时间点经过处理后的基因表达是否有差异。Hotelling T2检验可用来验证随着时间的推移,基因的表达谱是否发生变化。随着时间的进程,某些基因的表达上调或下调,某些基因的表达保持不变,可以发现随着时间及条件的变化表现出不同表达水平的标识基因。基因表达模式的时间序列也有利于理解与疾病相关的表达通路及其功能。单样本t检验可用于评估在每个特定的时间点基因的表达是否上调和下调或保持不变。那么,基因表达模式的时间进程可通过一类分类技术来加以分析,这对理解基因间的调控过程和生物通路提供了非常有用的工具[15]。Schulte 等[18]研究了不同的基因表达模式,包括即早基因、“延迟”基因和效应基因在神经母细胞瘤中TrkA和TrkB 受体中的表达情况,进而发现在诱导即早基因和下游的靶点调控中的分子机理。
目前,在时间序列微阵列研究中已发展了许多方法,这些方法都是针对提取时间过程中的差异表达基因。Storey等[19]利用基于spline的方法发现了在时间过程中基因表达的改变。Yuan 等[20-21]分别在2006和2008 年用基于隐马尔科夫算法的模型分析了多种生物条件下的时间序列微阵列数据。Tai和Speed[22]提出了一种多变量经验贝叶斯的统计方法鉴别差异表达基因。Ma 等[23]在2009 年通过功能性ANOVA 混合效应模型将时间序列基因表达值分类,并且鉴别差异表达基因。Zhou 等[24]在2010 年根据时间序列基因表达数据发展了一种对生存结果的预测模型。Tibshirani 等[25]在2013 年提出了一种根据时间序列基因表达谱对病人分类的方法,这种方法的提出为肿瘤的个性化治疗提供了新的手段。
基于通路和GSEA 的方法在功能基因组学研究中已经发展了十余年。由于不完整的信息和通路数据注释不佳,研究人员开始结合基因集富集分析方法和基于网络模块的方法,以鉴别较大幅度的分子机制。第三代基因表达谱分析方法(包括基因集/通路/网络分析)可被定义为一个以知识为导向的数据驱动的方法,这不仅是基于先验基因集的知识,而且利用了基因集内部或基因集之间的通路/网络的拓扑结构。2007 年,Vidal[26]的研究小组在哈佛大学利用各种生物信息学数据集对乳腺癌的易感性构建蛋白质相互作用网络,并确定HMMR 为新的疾病易感位点。随后,TreyIdeker[27]的研究小组在加州大学圣地亚哥分校综合蛋白质网络和基因表达数据,以提高乳腺癌患者转移形成的预测。这2 项研究是一个新的里程碑,标志着网络和通路的激动人心的开始,虽然容易出错,而且不完整,但可作为一种导向,引导今后的微阵列数据分析。
基因芯片技术已被广泛应用于遗传变异,基因网络、调控过程中的相互作用,以及生物通路等方面的研究,已成为理解基因的相互作用、协同、网络调控等的有力工具[28]。
7 结语
微阵列技术正深入到人类肿瘤疾病研究的各个方面。与其他研究方法相比,该技术更关注肿瘤在不同条件下基因表达的变化。可以通过微阵列技术对基因组进行分析来确定新的潜在的治疗途径或研发新的诊断试剂,即所谓的生物标志物发现研究[29]。
由于肿瘤受多重因素的影响,因此我们所获得的差异表达基因,哪怕是一个简单的比较试验,也可能受到其他信号的干扰。癌症样本的来源各异,包括直接手术活检的样本、很有限的针刺活检样本、尸检样本、特定癌症所建立的细胞系,甚至是石蜡固定样本的切片。当比较肿瘤样本及其正常对照时,必须确保样本的匹配度,如乳腺癌样本必须与相对应的正常乳腺细胞相比较。但这可能很难做到,如活检样本不可能是均质的,含有多种细胞类型,是正常和恶性细胞或不同阶段肿瘤细胞的混合物。此外,肿瘤细胞的正常对照也可能并不确定,因此需要分析多个不同的正常样本。同样,不同患者样本的遗传差异也可能会影响结果,需要增加足够的对照以减少这些因素的干扰。显然,正常样本和肿瘤样本的匹配程度也取决于实验的目标。如探索性分析实验的目的是了解某一系统的基本生物学特征,那么样本匹配度的要求可能不像生物标志物筛选那么严格,因为生物标志物筛选的目标是确定可靠的诊断工具。因此,实验设计、原始数据分析及统计方法的选择,是肿瘤研究中至为重要的步骤。
将一个基因表达谱或特殊基因表达信号转换成生物学上可以理解的概念,仍然是一个需要很大努力和充满挑战的任务。在这方面,对人类基因组及其他模式生物基因组功能越来越多的了解,将为基因表达研究提供大量的补充信息。另外,近年出现的系统生物学,在转录组水平的目的是能够描述支持个体基因表达状态的基因调节网络,也将及时提供细胞转录水平的可预见的详细图谱。通过表达谱分析,可以预测疾病状态的细胞和组织中受影响的特殊生化途径和生物学过程。
肿瘤基因表达谱数据挖掘不仅对认识肿瘤发生发展的机理具有重要意义,而且也会为肿瘤的分子诊断和防治开辟全新的途径,并有助于肿瘤个性化治疗的实现。利用基因表达谱对肿瘤样本进行准确诊断,构建肿瘤基因调控网络,是一项具有重要意义的大课题。
[1]Dudoit S,Yang Y,Callow M J,et al.Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments[J].Stat Sin,2002,12:111-139.
[2]Tseng G C,Oh M K,Rohlin L,et al.Issues in cDNA microarray analysis:quality filtering,channel normalization,models of variations and assessmentof gene effects[J].Nucleic Acids Res,2001,29(12):2549-2557.
[3]Fan J,Tam P,Vande Woude G,et al.Normalization and analysis of cDNA micro-arrays using within-array replications applied to neuroblastoma cell response to a cytokine[J].Proc Natl Acad Sci USA,2004,101(5):1135-1140.
[4]Fan J,Peng H,Huang T.Semilinear high-dimensional model for normalization of microarray data:a theoretical analysis and partial consistency[J].J Am Stat Assoc,2005,100(471):781-813.
[5]Huang J,Wang D,Zhang C H.A Two-way semi-linear model for normalization and analysis of cDNA microarray data[J].J Am Stat Assoc,2005,100:814-829.
[6]Ma S,Kosorok M R,Huang J,et al.Robust semiparametric cDNA microarray normalization and significance analysis[J].Biometrics,2006,62(2):555-561.
[7]Cui X,Hwang J T G,Qiu J,et al.Improved statistical tests for differential gene expression by shrinking variance components estimates[J].Biostatistics,2005,6(1):59-75.
[8]Smyth G K,Michaud J,Scott H S.Use of within-array replicate spots for assessing differential expression in microarray experiments[J].Bioinformatics,2005,21(9):2067-2075.
[9]Kerr M K,Churchill G A.Experimental design for gene expression microarrays[J].Biostatistics,2001,2(2):183-201.
[10]Tusher V G,Tibshirani R,Chu G.Significance analysis of microarrays applied to the ionizing radiation response[J].Proc Natl Acad Sci USA,2001,98(9):5116-5121.
[11]Storey J D,Tibshirani R.Statistical significance for genomewide studies[J].Proc Natl Aca Sci USA,2003,100(16):9440-9445.
[12]Dudoit S,Shaffer J P,Boldrick J C.Multiple hypothesis testing in microarray experiments[J].Stat Sci,2003,18(1):71-103.
[13]Inamura K,Fujiwara T,Hoshida Y,et al.Two subclasses of lung squamous cell carcinoma with different gene expression profiles and prognosis identified by hierarchical clustering and non-negative matrixfactorization[J].Oncogene,2005,24:7105-7113.
[14]Hastie T J,Tibshirani R,Friedman J.The elements of statistical learning:data mining,inference and prediction[M].2nd ed.New York:Springer,2005:83-85.
[15]Svrakic N M,Nesic O,Dasu M R K,et al.Statistical approach to DNA chip analysis[J].Recent Prog Horm Res,2003,58:75-93.
[16]Tibshirani R,Hastie T,Narasimhan B,et al.Diagnosis of multiple cancer types by shrunken centroids of gene expression[J].Proc Natl Acad Sci USA,2002,99(10):6567-6572.
[17]Fan J,Li R.Statistical challenges with high dimensionality:feature selection in knowledge discovery[J].Proc Madrid Intl Congress Math,2006,111:595-622.
[18]Schulte J,Schramm A,Klein-Hitpass L,et al.Microarray analysis reveals differential gene expression patterns and regulation of single target genes contributing to the opposing phenotype of TrkA-and TrkB-expressing neuroblastomas[J].Oncogene,2005,24(1):165-177.
[19]Storey J D,Xiao W J,Tompkins R,et al.Significance analy-sis of time course microarray experiments[J].Proc Natl Acad Sci USA,2005,102(36):12837-12842.
[20]Yuan M,Kendziorski C.Hidden markov models for microarray time course data under multiple biological conditions[J].J Am Stat Assoc,2006,101(476):1323-1332.
[21]Yuan Y,Li C T,Wilson R.Partial mixture model for tight clustering of gene expression time-course[J].BMC Bioinf,2008,9:287.
[22]Tai Y C,Speed T P.A multivariate empirica bayes statistic for replicated microarray time course data[J].Ann Stat,2006,34(5):2387-2412.
[23]Ma P,Zhong W,Liu J S.Identifying differentially expressed genes in time course microarray data[J].Stat Biosci,2009,1:144-159.
[24]Zhou B,Xu W,Herndon D,et al.Analysis of factorial timecourse microarrays with application to a clinical study of burn injury[J].Proc Natl Acad Sci USA,2010,107(22):9923-9928.
[25]Zhang Y,Tibshirant R,Davis R.Classification of patients from time-course gene expression[J].Biostatistics,2013,14(1):87-98.
[26]Pujana M A,Han J D J,Starita L M,et al.Network modeling links breast cancer susceptibility and centrosome dysfunction[J].Nat Genet,2007,39(11):1338-1349.
[27]Chuang H Y,Lee E,Liu Y T,et al.Network-based classification of breast cancer metastasis[J].Mol Syst Biol,2007,3:140-149.
[28]Akiyoshi T,Kobunai T,Watanabe T.Predicting the response to preoperative radiation or chemoradiation by a microarray analysis of the gene expression profiles in rectal cancer[J].Surgery Today,2012,42(8):713-719.
[29]Qian Z,Qingshan C,Chun J,et al.High expression of TNFSF13 in tumor cells and fibroblasts is associated with poor prognosis in non-small cell lung cancer[J].Am J Clin Pathol,2014,141(2):226-233.
猜你喜欢
环球时报(2022-09-29)2022-09-29
中学生数理化·高一版(2021年2期)2021-03-19
知识经济·中国直销(2018年8期)2018-08-23
数学学习与研究(2017年3期)2017-03-09
现代检验医学杂志(2016年3期)2016-11-15
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国老区建设(2016年1期)2016-02-28
湖北工业大学学报(2016年5期)2016-02-27
物理实验(2015年9期)2015-02-28
河南科技(2014年10期)2014-02-27
