
环孢菌素D衍生物PSC833逆转K562/DOX细胞的凋亡抗性及其机制
2014-08-13 12:12王建刚王淑英
中国病理生理杂志 2014年10期
刘 玲, 王建刚, 李 艳, 王淑英
(河南科技大学医学院药学系,河南 洛阳 471003)
多药耐药性(multidrug resistance, MDR)是肿瘤化疗失败的重要原因,P-糖蛋白(P-glycoprotein,P-gp)的高表达是肿瘤MDR的重要机制之一。P-gp为mdr1a基因的产物,由1 280个氨基酸组成,分子量170~180 kD,属于能量依赖性药物外排泵,可将抗癌药物由肿瘤细胞内外排至细胞外,使肿瘤细胞内药量减少,引起MDR的产生[1]。P-gp逆转剂都是P-gp底物,可竞争性或非竞争性抑制P-gp的外排功能,从而增加肿瘤细胞内药物蓄积,达到逆转肿瘤耐药的目的。PSC833是环孢菌素D的衍生物,具有非常强的P-gp逆转作用,同时没有环孢菌素A(cyclosporine A, CsA)的免疫抑制作用,是近年来发现的一种较好的P-gp抑制剂,也是目前唯一的进行临床研究的P-gp逆转剂。体外试验研究表明,PSC833对P-gp的抑制作用是CsA的10~20倍,对P-gp的结合力也比CsA高,因而不易被P-gp泵出到细胞外[2]。本实验拟观察环孢菌素D衍生物PSC833能否逆转人白血病细胞耐药株K562/DOX的抗凋亡作用,以及与凋亡调控蛋白表达的关系,深入探讨其逆转多药耐药的分子机制。
材 料 和 方 法
1 材料与细胞培养
PSC833由中国药科大学何玲教授提供;RPMI-1640培养基和胎牛血清(Gibco);MTT (Sigma);Annexin V/PI双染试剂盒(BD);盐酸阿霉素(浙江海正药业股份有限公司)、硫酸长春新碱(上海华联制药有限公司);活性氧(DCFH-DA)检测试剂盒和线粒体膜电位检测试剂盒(JC-1)、Fluo-3/AM、细胞裂解液、BCA蛋白含量检测试剂盒、线粒体分离试剂盒(碧云天生物技术研究所);细胞周期测定试剂盒、ECL检测试剂盒(南京凯基生物技术有限公司);细胞色素 C(cytochrome C, Cyt C)、Bax、Bcl-2、cleaved caspase-3、β-actin抗体和anti-rabbit IgG购于Bioworld。其余试剂均为市售分析纯。
人白血病细胞系K562及其耐药细胞系K562/DOX(耐阿霉素)、人脐静脉血管内皮细胞(human umbilical vein endothelial cells, HUVECs)由本室保存和提供。K562/DOX、K562和HUVECs用含10%小牛血清的RPMI-1640 培养基在37 ℃、5% CO2、饱和湿度的条件下培养,在K562/DOX细胞培养基中加入0.5 μmol/L阿霉素,实验前14 d更换无阿霉素的培养基后继续培养。
2 方法
2.1MTT法测定PSC833的耐药逆转实验 收集对数生长期K562/DOX和K562细胞,以5×104cells/well的密度接种于96孔板,24 h后加入不同浓度的DOX/VCR,检测在有或无PSC833存在条件下,DOX/VCR对细胞的毒性。PSC833终浓度为1、 2.5、 5和10 μmol/L。空白对照组加入等体积的PBS。每一浓度设4个平行孔。待细胞与药物作用44 h后,每孔加入5 g/L MTT,继续培养4 h,加入细胞裂解液,静置过夜使细胞裂解结晶全部溶出,用酶标仪在570 nm处测定吸光度(A),按以下公式计算抑制率,并求出半数抑制率(IC50)和逆转倍数(reversal fold, RF)。抑制率(%)=(1-实验组A/空白对照组A)×100%;RF= IC50(单用细胞毒药物)/ IC50(细胞毒药物+P-gp逆转剂)。
同时,采用MTT法检测PSC833对HUVECs的毒性作用,方法同上。
2.2PI染色检测细胞周期 将对数生长期的细胞消化接种到培养瓶中,次日,待细胞贴壁后,根据组别设置加入相应的含药培养基,同时设立阴性对照组、PSC833单用组(10 μmol/L)、DOX单用组(10 μmol/L)、DOX(10 μmol/L)和PSC833(2.5 μmol/L、5 μmol/L和10 μmol/L)合用组;药物作用24 h后,用0.25%胰酶(不含EDTA)消化收集细胞;用PBS洗涤细胞2次(2 000 r/min 离心5 min),收集并调整细胞浓度为1×109/L;制备的单细胞悬液用体积分数为70%乙醇固定2 h,4 ℃保存,染色前用PBS洗去固定液;加100 μL RNase A 37 ℃水浴30 min;再加入400 μL PI染色混匀,4 ℃避光30 min;上机检测,记录激发波长488 nm 处红色荧光。
2.3AnnexinV/PI双染流式细胞术检测细胞凋亡 取对数生长期的K562/DOX细胞接种于6孔板中,分组同上,经PSC833 2.5、5和10μmol/L作用24h后,将收集的细胞用冷PBS洗2次,缓冲液(bindingbuffer)用三蒸水稀释10倍,用缓冲液配成1×109/L的细胞浓度;吸取100μL至试管中,加入AnnexinV试剂和PI各5μL,混匀避光孵育15min;加入400μL染色缓冲液,混匀,流式细胞术检测。
2.4流式细胞术检测线粒体膜电位、活性氧水平和细胞内钙 取处于对数生长期状态良好的细胞,消化后接种于6孔板中,培养24 h,分组同上。换用含3%血清的RPMI-1640培养基,加入药物继续培养24 h。收集细胞,PBS洗涤2次,离心,弃上清,重悬于0.5 mL含3%血清的RPMI-1640培养基中。加入0.5 mL JC-1染色工作液,颠倒数次混匀。细胞培养箱中37 ℃孵育20 min。孵育结束后,600×g4 ℃离心3~4 min,弃上清,用JC-1染色缓冲液洗涤2次后,用适量JC-1染色缓冲液重悬后,用流式细胞术分析。
药物作用24 h后,细胞处理同上。按照1∶1 000用无血清培养液稀释DCFH-DA,使终浓度为10 μmol/L。加入适当体积稀释好的DCFH-DA。37 ℃细胞培养箱内孵育20 min。用无血清细胞培养液洗涤细胞3次,以充分去除未进入细胞内的DCFH-DA。用流式细胞术检测细胞的平均荧光强度。
药物作用24 h后,细胞处理同上。换用含3%血清的RPMI-1640培养基,加入药物继续培养24 h。收集细胞,PBS洗涤2次,离心,弃上清。加入5 μmol/L Fluo-3/AM,37 ℃孵育30 min,再次洗涤后,用流式细胞术分析。
2.5Westernblotting检测蛋白表达 加药处理细胞24h后,收集细胞并用裂解液裂解细胞获得总蛋白。采用BCA试剂盒于562nm处测定样品蛋白含量。然后,采用12%SDS-PAGE分离总蛋白,蛋白质转移到PVDF膜,5%脱脂奶粉室温封闭2h。封闭过的膜用TBST洗涤3次。将膜放入杂交袋中,加入Ⅰ抗4 ℃孵育过夜,使抗原抗体充分结合。隔天,将膜从杂交袋中取出,用TBST洗涤3次;再放入新杂交袋中,加入Ⅱ抗以结合Ⅰ抗,室温孵育膜2h。化学发光法检测,用凝胶成像系统拍照成像。目的蛋白的灰度值除以内参照β-actin的灰度值以校正误差,所得结果代表某样品的目的蛋白相对含量。
3 统计学处理
数据以均数±标准差(mean±SD)表示,用SPSS 13.0软件分析,组间均数的比较采用方差分析,以P<0.05为差异有统计学意义。
结 果
1 PSC833对K562/DOX细胞耐药的逆转作用
K562/DOX细胞对DOX和VCR的耐药倍数分别为52.44和564倍;PSC833与DOX/VCR合用后,IC50呈剂量依赖性减少,而RF呈剂量依赖性增加;对K562细胞,则不改变其IC50值,各组间差异无统计学意义,见表1、2。

表1 PSC833对K562/DOX细胞DOX的耐药逆转作用
*P<0.05,**P<0.01vsDOX.

表2 PSC833对K562和K562/DOX细胞VCR的耐药逆转作用
**P<0.01vsVCR.
PSC833在50 μmol/L以下时对HUVECs细胞的生长没有显著影响,细胞存活率在90%以上,见图1。
2 PSC833对DOX阻滞细胞周期的影响
与对照组相比,PSC833单用组和DOX单用组对细胞周期影响较小。而PSC833合用DOX可剂量依赖性增加G2/M期细胞的比例,减少G0/G1期细胞的比例,使细胞周期阻滞于G2/M期,对S期细胞的比例无明显改变,表明PSC833的耐药逆转作用与阻滞细胞周期G2/M期有关,见图2。
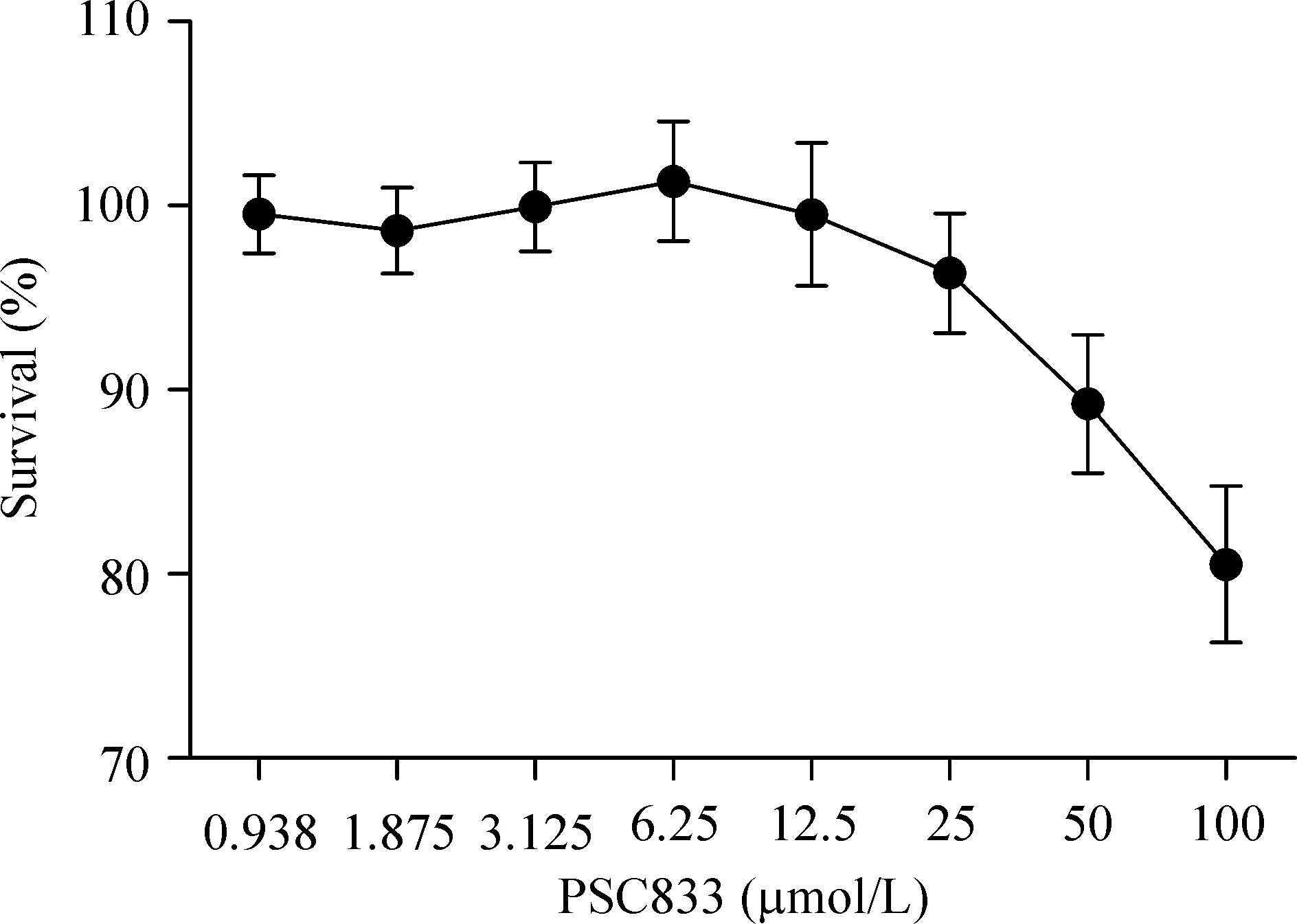
Figure 1. The effect of PSC833 on HUVEC growth.Mean±SD.n=3.
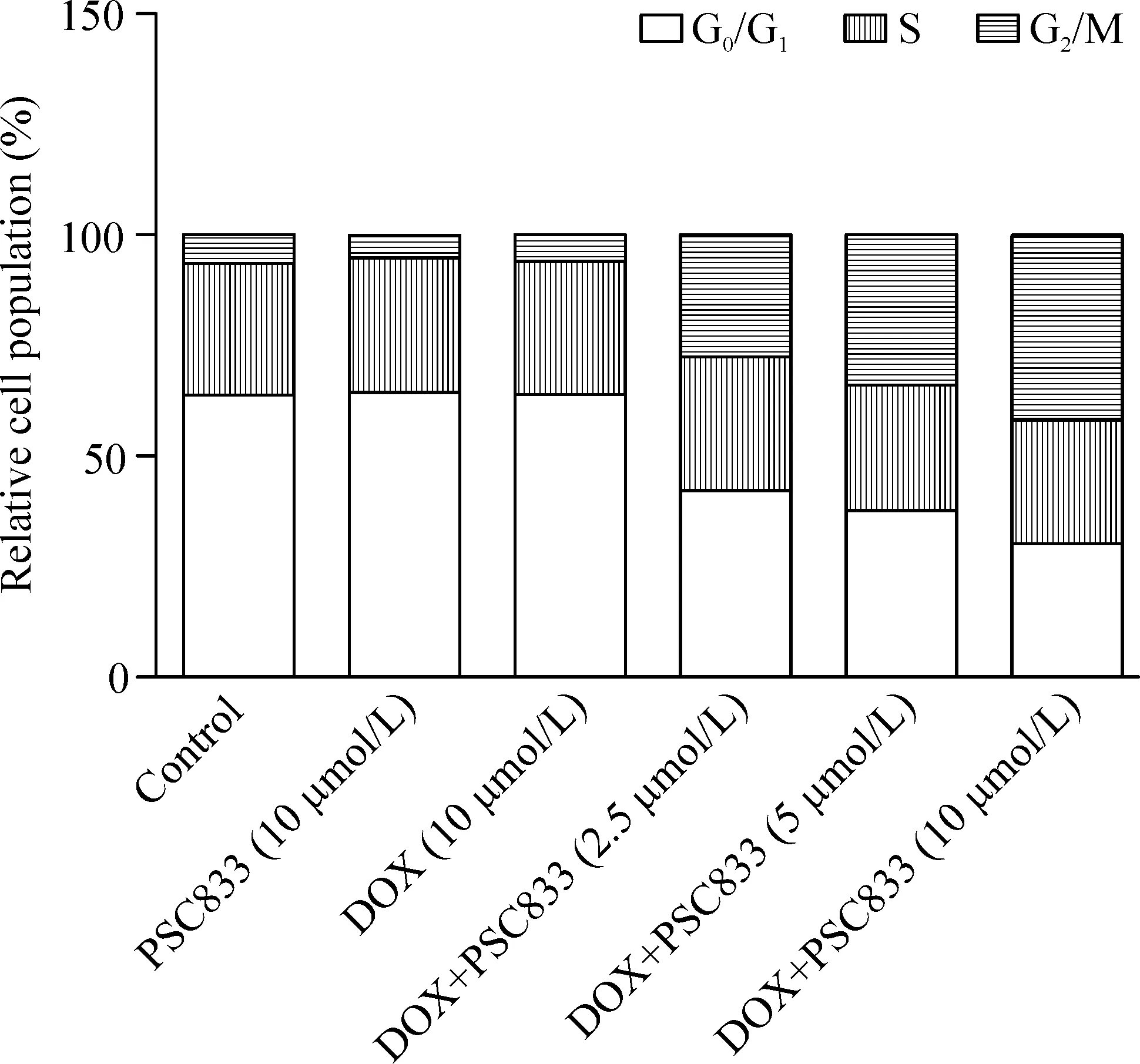
Figure 2. The effect of PSC833 on DOX-induced cell cycle progression in K562/DOX cells.
3 PSC833对DOX诱导线粒体膜电位的影响
与对照组相比,PSC833单用组和DOX单用组对线粒体膜电位影响较小。与DOX单用相比,PSC833和DOX合用后引起线粒体膜电位下降,Q4区比率明显增加,表明线粒体膜电位降低幅度加大,达到85.9%,见图3。
4 PSC833对DOX引起的活性氧水平的影响
与对照组相比,PSC833单用组和DOX单用组对活性氧水平影响较小。与DOX单用组相比,PSC833和DOX合用后可引起活性氧水平显著增加,见图4。
5 PSC833对DOX引起的细胞内钙的影响
PSC833和DOX合用后,K562/DOX细胞内的荧光强度明显升高,[Ca2+]i增加;单用PSC833和单用DOX细胞内[Ca2+]i均未见显著变化,见图5。
6 PSC833对DOX诱导的K562/DOX细胞凋亡的影响
与对照组相比,PSC833单用时对K562/DOX细胞凋亡率影响较小,DOX单用时对K562/DOX细胞凋亡率仅8.25%。随着PSC833浓度的增加,早期凋亡细胞、晚期凋亡和坏死细胞数量显著增加,且作用呈剂量依赖性,见图6。
7 PSC833对阿霉素诱导的K562/DOX细胞凋亡相关蛋白的影响
与对照组相比,PSC833和DOX单用时对凋亡蛋白Cyt C、Bcl-2、Bax和cleaved caspase-3表达影响较小;PSC833和DOX作用24 h后,线粒体中的Cyt C表达减少,而胞浆中Cyt C的表达水平相应增加;Bcl-2下调,Bax上调,caspase-3前体剪切形成活化的cleaved caspase-3,并且cleaved caspase-3的表达量随着PSC833的浓度增加而升高,诱导K562/DOX耐药细胞凋亡,见图7。

Figure 3. The effect of PSC833 on DOX-induced mitochondrial membrane potential in K562/DOX cells.Mean±SD.n=3.** P<0.01 vs DOX.

Figure 4. The effect of PSC833 on DOX-induced intracellular ROS production in K562/DOX cells. Mean±SD.n=3.** P<0.01 vs DOX.

Figure 5. The effect of PSC833 on DOX-induced intracellular calcium fluorescent intensity in K562/DOX cells. Mean±SD.n=3.*P<0.05, ** P<0.01 vs DOX.
讨 论
关于MDR产生的机理目前还不特别明确,但大多数具MDR的细胞膜表面都有P-gp的过度表达。K562/DOX耐药细胞系是由K562细胞经过长期的阿霉素诱导和筛选而建立起来的典型的耐药细胞系,mdr1 mRNA及P-gp高表达,不仅对诱导药物阿霉素有很强的耐药性,而且对抗肿瘤药物也具有交叉耐药性。PSC833作为多药耐药逆转剂,能增强MDR细胞系对其它化疗药物如阿霉素、紫杉醇等的敏感性。研究发现,PSC833可增加耐阿霉素的肺癌细胞株(SK-MES-1/DX1000)对阿霉素的敏感性,降低P-gp底物Rh123在细胞内的累积,下调MDR1 mRNA和P-gp的表达,激活JNK/c-Jun/AP-1通路和抑制NF-κB的表达,从而逆转耐药细胞株(SK-MES-1/DX1000)对阿霉素的耐药性[3]。
肿瘤的多药耐药性与抗凋亡作用关系密切。研究发现,阿霉素可激活细胞内酸性鞘磷脂酶,导致神经酰胺形成,最终激活caspase-3而引起细胞凋亡,caspases的活化及其所引发的细胞毒作用也需要酸性的细胞内环境[4]。作为抗凋亡分子,P-gp可跨膜转运内源性鞘磷脂,通过减少质膜上的鞘磷脂使神经酰胺生成减少,阻止凋亡的发生;P-gp还可通过增加细胞内pH, 降低细胞内游离药物浓度,并使caspases处于失活状态,从而抑制caspases依赖性的细胞凋亡[5]。线粒体膜电位的丧失是线粒体凋亡途径的先决条件。当线粒体内活性氧大量产生,线粒体膜通透性改变,导致线粒体膜势能发生改变,进而引起线粒体释放Cyt C及细胞凋亡起始因子(apoptosis initiating factors, AIFs)等,这样可以激活caspases,从而诱导细胞凋亡的发生[6]。另外,线粒体外膜通透性的改变也可直接由Bcl-2家族蛋白控制。Ca2+作为第二信使触发或者调控多种细胞内事件而使细胞执行一系列特定的功能。钙离子不仅参与细胞凋亡早期Cyt C的释放和caspase-3的激活,而且也作用于凋亡晚期的DNA裂解[7]。
研究发现,PSC833能显著抑制P-gp的外排功能,增加耐药细胞对其它药物的敏感性,逆转细胞的多药耐药性[8-9]。但PSC833在DOX诱导细胞凋亡过程中所致的活性氧与线粒体膜电位的改变均未见报道。因此,我们进一步观察了PSC833对K562/DOX细胞阿霉素、长春新碱耐药的逆转作用,及其对K562/DOX细胞凋亡和相关蛋白表达的影响。结果显示,PSC833可剂量相关性地逆转K562/DOX细胞对DOX/VCR的耐药、明显增加DOX诱导的K562/DOX细胞凋亡。本研究利用荧光探针DCFH-DA检测到PSC833和DOX处理后,K562/DOX细胞活性氧明显增加,说明PSC833诱导细胞凋亡的过程中活性氧的改变起到了重要的作用。活性氧的改变可导致线粒体膜电位的改变,因此我们利用JC-1荧光探针,用流式细胞术检测细胞线粒体膜电位水平,发现耐药细胞由Q2向Q4象限移动(膜电位降低),呈一定的剂量依赖性,证实PSC833和DOX合用可降低线粒体膜电位,由此推测PSC833的耐药逆转作用可能是通过线粒体通路诱导细胞凋亡发挥抗肿瘤作用的。[Ca2+]i的升高被认为是凋亡的启动环节。结果显示,K562/DOX细胞内的荧光强度明显升高,[Ca2+]i显著增加,因此Ca2+作为第二信使也参与了凋亡过程。线粒体外膜的损伤可以导致Cyt C释放,与Apaf-1结合后,形成一个多聚体即凋亡体,凋亡体募集pro-caspase-9,使其自我激活。Caspase-9前体聚合后被反式催化激活,活化的caspase-9继而激活下游执行caspases(caspase-3、-6、-7),切割许多重要底物,导致凋亡的发生。本研究发现PSC833和DOX合用后,线粒体中Cyt C表达降低而胞浆中表达升高,提示凋亡线粒体通路的激活;同时,抗凋亡蛋白Bcl-2表达下调,促凋亡蛋白Bax表达上调,激活cleaved caspase-3而启动了级联反应,诱导细胞凋亡。PSC833对K562/DOX细胞耐药逆转作用可能是PSC833抑制P-gp的外排作用,使细胞内DOX浓度增加,增强了DOX的凋亡诱导作用;另一方面,可能降低了P-gp对细胞凋亡的抑制作用,使K562/DOX细胞更易发生凋亡,从而介导活性氧升高,导致细胞线粒体膜电位的降低、[Ca2+]i显著增加,释放Cyt C,激活caspase-3,导致细胞凋亡。

Figure 6. The effect of PSC833 on DOX-induced apoptosis in K562/DOX cells.Mean±SD.n=3. **P<0.01 vs DOX.
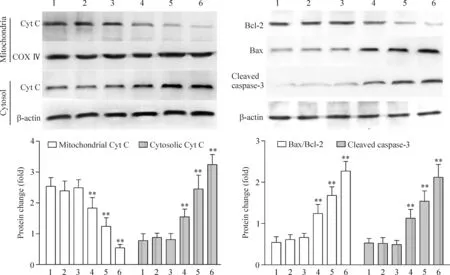
Figure 7. The effect of PSC833 on DOX-induced expression of Cyt C, Bcl-2, Bax and cleaved caspase-3 proteins.1: control; 2: DOX (10 μmol/L); 3: PSC833 (10 μmol/L); 4: PSC833 (2.5 μmol/L)+DOX; 5: PSC833 (5 μmol/L)+DOX; 6: PSC833 (10 μmol/L)+DOX.Mean±SD.n=3.** P<0.01 vs 2.
[参 考 文 献]
[1]BreierA,GibalovaL,SeresM,etal.Newinsightintop-glycoproteinasadrugtarget[J].AnticancerAgentsMedChem, 2013, 13(1):159-170.
[2] Zhang Q, Li F. Combating P-glycoprotein-mediated multidrug resistance using therapeutic nanoparticles [J]. Curr Pharm Des, 2013, 19(37):6655-6666.
[3]BarkH,ChoiCH.PSC833,cyclosporineanalogue,downregulatesMDR1expressionbyactivatingJNK/c-Jun/AP-1andsuppressingNF-kappaB[J].CancerChemotherPharmacol, 2010, 65(6):1131-1136.
[4] Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives [J]. Curr Cancer Drug Targets, 2013, 13(3):326-346.
[5]GibalováL,SerešM,RusnákA,etal.P-glycoproteindepressescisplatinsensitivityinL1210cellsbyinhibitingcisplatin-inducedcaspase-3activation[J].ToxicolIn Vitro, 2012, 26(3):435-444.
[6] Indo HP, Davidson M, Yen HC, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage [J]. Mitochondrion, 2007, 7(1-2):106-118.
[7] 卫小红,邵 杰,王军辉. 齐墩果酸诱导人肺腺癌细胞A549凋亡及其与细胞内钙离子的关系[J]. 同济大学学报(医学版), 2009, 30(5):19-23.
[8] Nagao K, Maeda M, Maucat NB, et al. Cyclosporine A and PSC833 inhibit ABCA1 function via direct binding [J]. Biochim Biophys Acta, 2013, 1831(2):398-406.
[9]MyllynenP,KurttilaT,VaskivuoL,etal.DNAdamagecausedbybenzo(a)pyreneinMCF-7cellsisincreasedbyverapamil,probenecidandPSC833[J].ToxicolLett, 2007, 28, 169(1):3-12.
猜你喜欢
现代农业科技(2022年5期)2022-12-14
科学导报(2022年11期)2022-03-03
世界科学技术-中医药现代化(2021年7期)2021-11-04
智慧健康(2020年9期)2020-12-03
肿瘤防治研究(2019年7期)2019-08-01
教育教学论坛(2019年19期)2019-06-17
中国中药杂志(2017年11期)2017-06-22
中国计划生育学杂志(2017年3期)2017-06-01
郑州大学学报(医学版)(2015年1期)2015-02-27
中国药业(2014年24期)2014-05-26
