
纤维素选择性催化转化为重要平台化合物的研究进展
2013-10-11 08:34郭庆祥
化工进展 2013年2期
邓 理,廖 兵,郭庆祥
(1中国科学院纤维素化学重点实验室,广州化学研究所,广东 广州 5106502;2中国科学技术大学化学系,安徽省生物质洁净能源重点实验室,安徽 合肥 230026)
在资源逐渐匮乏的今天,可持续发展成为人们追求的目标,生物质资源替代化石资源正得到越来越多的关注。纤维素作为自然界中总量最多的植物生物质,已经在人类社会的发展过程起到了重要的作用,如利用纤维素生产纸张和棉织物。通过化学催化方法,纤维素还可以转化为人类需要的多种材料、燃料以及化学品。最新的研究显示通过水解、加氢、热解和脱水等反应可以有效地转化纤维素制备葡萄糖、山梨糖醇、乙二醇、合成气、芳香烃以及呋喃化合物。上述化合物被称为未来生物精炼的“积木”(building blocks)[1-2]。
2004年,美国能源部发布的一份名为“源自生物质的高附加值化学品”报告首次提出了12种来源于碳水化合物的平台化合物,并将其形象地称为生物精炼的“积木”,意在说明生物质通过生物或化学的转化可以有效地获得这些化合物,再通过这些“积木”构建出更多的化合物,最终成为人们需要的药物、精细化学品、材料、燃料等。这12种平台化合物包括:丁二酸、2,5-呋喃二酸、3-羟基丙酸、天冬氨酸、葡萄糖二酸、谷氨酸、衣康酸、乙酰丙酸、3-羟基丁内酯、甘油、山梨糖醇和木糖醇[1]。
本文作者将针对以纤维素为原料催化制备平台化合物的路线进行综述,重点介绍最近发展的纤维素化学催化转化的新方法和新路线。
1 纤维素化学结构简介
纤维素是自然界中含量最高的生物质组分(30%~50%)。在众多糖苷键中,纤维素的β-1,4-糖苷键最为稳定,再加上纤维素结构的特性使得大量β-1,4-糖苷难以被酶或者酸接触,更增加了纤维素水解的难度[3]。
纤维素是由β-D-葡萄糖通过β-1,4-糖苷键连接而成的线性规整的高分子,见图1。纤维素结构中除了具有β-1,4-糖苷键外还具有大量的氢键,纤维素的氢键一般分两种:分子内氢键和分子间氢键,即在一条纤维素链上相邻的葡萄糖单元形成的氢键和链与链之间形成的氢键。一般而言,聚合度在2~6的纤维素低聚物可以溶于水,聚合度在7~13的低聚物可以溶于热水,聚合度更高则不溶于水,而且聚合度高于30时,纤维素就会利用分子间氢键形成致密的结构[3-4]。天然植物中的纤维素的聚合度一般在1000以上,有的甚至高达上万,这使得植物能够抵御自然界中的化学和生物侵蚀,并且不溶于常规溶剂。

图1 纤维素结构示意图
纤维素按照晶体结构可以分为无定形和结晶纤维素,其中结晶纤维素按照晶型的不同又分为:Ⅰα、Ⅰβ、Ⅱ、Ⅲα、Ⅲβ、Ⅳα和Ⅳβ构型。其中,Ⅰα、Ⅰβ是天然纤维素的构型,藻类和细菌细胞壁中的纤维素以Ⅰα型为主,木材、棉花等植物的纤维素以Ⅰβ为主。需要注意的是,Ⅰα和Ⅰβ虽然是天然纤维素的构型,但它们并不是热力学最稳定的结构,热力学上能量有利的结构是Ⅱ构型。Ⅱ型纤维素可以通过Ⅰ型纤维素在浓氢氧化钠溶液中溶胀处理获得,也称丝光纤维素。由于葡萄糖单元的羟甲基可以围绕C5~C6键自由旋转,可以给出3种相对稳定的构象(图2),即tg、gg和gt。在Ⅰ型纤维素中通常采取tg构象,这使得Ⅰ型纤维素能够形成分子内氢键 O2—H··O6,而Ⅱ型纤维素中采取 gt构象,则不能生成O2—H··O6氢键,这给Ⅱ型纤维素生成分子间氢键提供了条件。Ⅲα和Ⅲβ型纤维素可以分别通过Ⅰ和Ⅱ型纤维素经氨气爆破处理获得,而Ⅳα和Ⅳβ型纤维素则可以通过Ⅲα和Ⅲβ型纤维素在甘油中加热到206 ℃获得。目前研究关注比较多的主要是Ⅰ和Ⅱ型纤维素[4]。
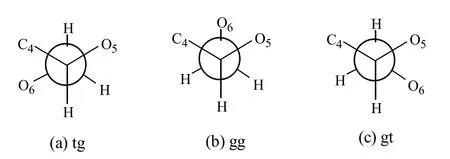
图2 葡萄糖单元构象
2 纤维素水解制备葡萄糖
一般而言,碳水化合物要进行生物转化都需要将其水解成水溶性的糖,然后再通过微生物发酵生成相应的产物,如谷氨酸、马来酸、天冬氨酸等。所以从这个角度,水溶性糖特别是葡萄糖是生物发酵过程的基础。
从纤维素得到葡萄糖可以通过两种途径:化学酸水解方法和生物酶水解方法。这里将介绍最近发展的酸催化纤维素水解的方法。目前,研究纤维素酸水解的新方法呈现出两大趋势:①利用离子液体溶解纤维素,形成均相溶液,再催化水解得到单糖或多糖;②在水中利用固体酸催化剂直接催化纤维素水解。
2.1 离子液体中催化纤维素水解
2002年,Rogers等[5]报道了利用离子液体溶解纤维素,纤维素浓度最大可达 25%(质量分数),这为纤维素后续均相转化奠定了基础。赵宗保等[6-7]首先发展了在离子液体中水解纤维素的方法,并将其应用到了木质纤维素的降解中(图3)。使用含有7%盐酸的[BMIM]Cl溶剂体系,在100 ℃反应6 h,玉米秆、稻草、松木和甘蔗渣的总还原糖产率分别为66%、74%、81%和68%。

图3 离子液体中无机酸催化纤维素水解
Raines等[8]在此基础上,通过多次分批加入水的方法,将纤维素制备葡萄糖的产率提高到了90%。通过使用离子排阻色谱可以分离离子液体和葡萄糖,再通过微生物发酵得到了乙醇,证明该过程生产的糖适合于生物发酵过程。类似的分离过程还可以通过中性氧化铝柱色谱实现[9]。
Schüth等[10-12]利用大孔阳离子树脂 Amberlyst 15DRY,在离子液体中催化纤维素水解,得到了纤维素的低聚物,聚合度约30,产率90%(图4)。他们指出在离子液体中得到的单糖很难高效地与离子液体分离,给后续利用带来困难。通过生成纤维素的低聚物,通过在离子液体中加入水则可以方便地将其析出,再利用纤维素酶可以更加容易地得到发酵所需的单糖,其优点在于方便了后续处理过程。

图4 离子液体中固体酸催化纤维素解聚
由于离子液体溶解过程破坏了纤维素的晶体结构,并且有效地降低了纤维素的聚合度,通过上述方法处理得到的纤维素低聚物能够很容易地被纤维素酶降解,酶水解的速度远远高于处理前的纤维素(2 h转化率90%vs20%)[11]。
2.2 固体酸催化纤维素水解
传统方法即利用稀酸水解纤维素,主要问题在于产生废酸和设备腐蚀等,使用固体酸可以针对性地解决上述问题。Onda等[13-14]研究发现使用磺化后的活性炭可以有效催化纤维素水解,效果优于其它固体酸,如 H-beta分子筛、γ-Al2O3、Amberlyst 15等。研究表明,经过球磨的纤维素(无定形结构的纤维素),能够在150 ℃、水解24 h后得到40%的葡萄糖,且催化剂能够重复使用。
Hara等[15-17]合成了一种结晶纤维素高温处理得到的炭负载的磺酸催化剂,该催化剂含有 1.9 mmol/g 磺酸根、2.0 mmol/g 羟基和0.4 mmol/g 羧基,比表面积只有2 m2/g。惊奇的是,该催化剂在100 ℃能够将微晶纤维素完全转化为水溶性的糖(图5)。通过MALDI-TOF-MASS检测产物发现,产物中含有葡萄糖、纤维二糖和大量的纤维多糖。动力学测试的结果表明,使用该催化剂催化纤维素水解的表观活化能是110 kJ/mol,远远小于硫酸催化该反应的表观活化能(170 kJ/mol)。该催化剂的良好性能可能来自于其对多糖的良好吸附作用。当纤维素完全转化后,该催化剂可以从反应体系中分离,从而实现催化剂重复使用。该催化剂使用 25次后未发现有失活现象。
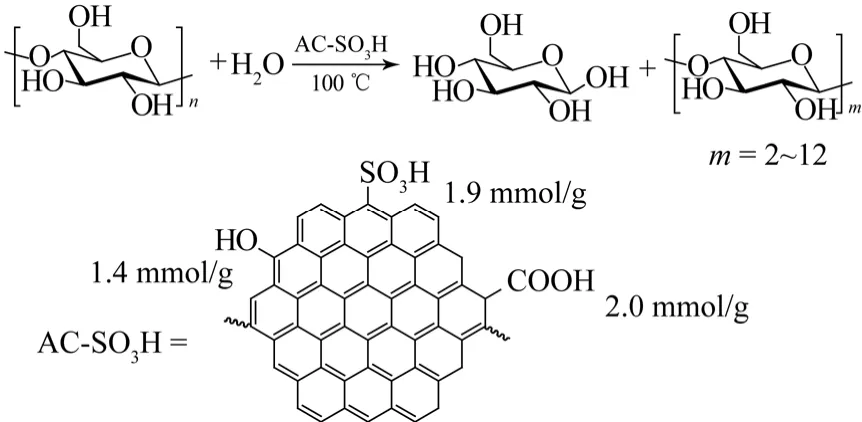
图5 磺化活性炭催化纤维素水解
张涛等[18]利用具有介孔结构的碳材料 CMK-3作为载体,合成了一种碳负载的磺酸型催化剂,成功地催化纤维素水解,纤维素转化率高达94%,葡萄糖产率75%。对比试验表明使用CMK-3作为载体催化效果明显优于其它碳载体。
需要指出的是,即使纤维素能够完全反应,形成均相的溶液,但对于真实生物质而言,其所含的木质素不能够被水解,仍然以固体形式存在,给催化剂的分离造成困难。为此,来大明等[19]合成了一种具有介孔结构的二氧化硅基固体酸,并在该固体酸中引入磁性四氧化三铁纳米颗粒,成功地实现了纤维素到葡萄糖的转化,葡萄糖产率达到50%,反应过后催化剂能够通过磁场方便地回收,实现了催化剂与产物和反应物的分离,且能够重复使用。另外,该催化剂还能够在较高的固液比条件下使用,可以有效减少后续处理过程中的能耗。
3 纤维素制备呋喃类化合物
3.1 羟甲基糠醛(HMF)的选择性合成
以葡萄糖为原料制备 HMF不仅要进行脱水,还要在脱水前实现异构化,即完成吡喃糖到呋喃糖的转化。CrCl2或CrCl3可以在离子液体中很好地完成这一任务,同时CrCl2或CrCl3作为路易斯酸也能够实现脱水反应,HMF的产率高达70%[20]。使用大位阻卡宾作为配体,优化该反应可将 HMF的产率提高到80%以上[21]。最近,利用拓展的X射线精细吸收谱(EXAFS)分析葡萄糖CrCl2在离子液体中的相互作用,发现起到异构化作用的是Cr离子的二聚体 Cr2Cl5−[22]。另外,韩布兴等[23]发现 SnCl4也有与 CrCl3类似的作用,能够催化葡萄糖到果糖的异构化。
在纤维素制备HMF方面,使用DMAc/ LiCl/HCl/CrCl3/离子液体多组分的纤维素溶剂体系,可以很好地将纤维素转化为HMF,产率54%[24]。基于离子液体/CrCl3体系,通过加入一定量的CuCl2,可以有效降低该体系的熔化温度和熔化热,表明两种金属的同时加入可以有效地破坏离子液体的长程相互作用。在离子液体/CrCl3/CuCl2反应体系中,纤维素可以在较低温度(120 ℃)下很好地完成解聚、异构化和脱水反应,得到55%的HMF[25]。
3.2 HMF的应用

图6 由HMF制备液态烷烃的路线
2005年,Dumesic等[26]率先提出利用HMF作为中间体由碳水化合物制备液态烷烃的思路(图6)。在他们以往的工作中,通过山梨糖醇重整获取烷烃只能得到含6个碳原子以下的烷烃,不能满足内燃机燃料的要求[27]。而利用HMF为原料,与另一分子丙酮发生羟醛缩合反应得到了具有9个碳原子的含氧中间产物,再通过加氢还原完全脱除分子中的氧,就可以高选择性地获得壬烷和庚烷,它们可以作为汽油的主要成分使用。上述羟醛缩合的反应条件为摩尔比110∶的HMF与丙酮水溶液在MgO/Al2O3催化下,室温反应5 h。如果HMF与丙酮的摩尔比为11∶,则烷烃产物以十四烷和十五烷为主,可以用作柴油。类似的反应也可以在丙酮和糠醛之间或丙酮、糠醛和 HMF三者之间发生,得到不同碳链长度的烷烃。
二甲基呋喃是一种潜在的燃料分子,由于含氧量低,其热值高达30 MJ/L。二甲基呋喃可以通过HMF在220 ℃、铜-钌催化剂的作用下加氢还原得到,产率达到71%。在制备过程中,由于二甲基呋喃水溶性差(水中溶解度为2.3 g/L),可以自动地与水分离,有助于节省分离过程的能耗[28]。
呋喃二甲酸具有与对苯二甲酸类似的结构,所以该分子被预测有代替对苯二甲酸生产 PET聚酯材料的巨大潜力。呋喃二甲酸的合成主要通过HMF的氧化获得。在强碱性条件下,使用贵金属催化剂,空气氧化HMF,呋喃二甲酸的选择性可高达99%。利用氧化对二甲苯生产对苯二甲酸的金属溴化物催化剂,如溴化钴、溴化锰和溴化锆,氧化 HMF制备呋喃二甲酸,产率也可达60%。通过一锅法合成呋喃二甲酸,也得到了研究人员的关注。利用果糖为原料在DMSO中,以磷钒酸为催化剂,可以得到81%的呋喃二甲酸。使用硅胶负载的钴催化剂,也可以催化该反应,呋喃二甲酸的选择性高达99%,果糖转化率72%[2]。
由 HMF还原得到的 2,5-二羟甲基呋喃是重要的药物合成中间体,以及冠醚和高分子的中间体。2,5-二羟甲基呋喃可以通过硼氢化钠还原 HMF获得,也可以利用H2还原得到,显然前者更适合实验室级别的合成,而后者对于规模较大的高分子材料生产更有吸引力。对 HMF加氢时,催化剂有很大的选择余地,例如:铜/铬催化剂、氧化钴、氧化铂、氧化钼、兰尼镍都可以催化该反应。在 140 ℃、7 MPa H2和铂催化剂的条件下,HMF能够完全转化为2,5-二羟甲基呋喃[29-30]。
3.3 氯甲基糠醛的合成
2008年,Mascal等[31]报道了纤维素高效转化的最新研究成果,他们使用浓盐酸和 5%氯化锂作为催化剂将纤维素高产率地转化为氯甲基糠醛,反应温度65 ℃,使用1,2-二氯乙烷连续萃取18 h,氯甲基糠醛的分离收率可达71%(图7)。在后续的工作中[32],他们通过使用浓盐酸/1,2-二氯乙烷两相体系提高了纤维素转化的效率,100 ℃下反应3 h,氯甲基糠醛产率可达80%以上。

图7 纤维素制备氯甲基糠醛及其衍生物
但需要注意的是,该方法的缺点在于:使用浓盐酸作为催化剂和溶剂,腐蚀性强需要特殊的反应装置;反应过程中使用到了有机溶剂 1,2-二氯乙烷作为萃取剂;每生产一分子氯甲基糠醛就消耗一个氯原子,而后续的处理中氯原子被取代,成为废弃物,不符合原子经济性。
3.4 氯甲基糠醛的应用
通过后续工作将其转化成为不含氯的呋喃衍生物。如使用氯化钯作催化剂对其催化加氢可以得到甲基糠醛,产率88%;使用乙醇室温搅拌8 h可以得到乙氧基甲基糠醛,产率95%,该化合物是一种潜在的内燃机燃料;通过水解还可以得到 HMF和乙酰丙酸[33]。
4 纤维素制备乙酰丙酸及其衍生物
4.1 乙酰丙酸的合成
由纤维素制备乙酰丙酸通常需要使用无机强酸作为催化剂。纤维素在酸的作用下水解得到葡萄糖,然后葡萄糖异构化并脱水生成 HMF中间体,HMF在高温酸性水溶液中不稳定,继续发生重排反应得到乙酰丙酸和副产物甲酸,反应机理见图8。

图8 HMF在水中降解的途径
美国 Biofine公司[34]发展了一种利用纤维素原料制备乙酰丙酸的连续生产工艺。首先,原料与2%~5%的硫酸水溶液混合进入第1个管式反应器,在215 ℃、3.1 MPa条件下反应15 s,从而将纤维素水解成单糖,再脱水生成HMF。含有HMF的反应液继续进入第2个反应器,在193 ℃、1.5 MPa下反应12 min,最终乙酰丙酸的产率可达70%。目前该公司的上述工艺已经建立了一套示范装置,并连续运行1年以上,每天可以处理纸浆1 t(干重),产出0.5 t乙酰丙酸以及副产物甲酸和糠醛。该公司预计1000~2000 吨/天装置建成后,乙酰丙酸的生产成本可以降低到0.09~0.11 美元/千克[35]。
最新的研究表明,在甲醇中使用混合酸或者固体酸也能够将纤维素转化为乙酰丙酸甲酯。由于在甲醇介质中,中间体糖苷、缩醛化的 HMF不易生成胡敏素,所以乙酰丙酸甲酯的产率更高[36-37]。
4.2 乙酰丙酸制备燃料
通过乙酰丙酸还原得到的γ-戊内酯可以作为汽油和柴油添加剂使用(图9),与汽油混合时其抗爆性与乙醇相同。工业上γ-戊内酯通过使用H2在非均相贵金属催化剂作用下对乙酰丙酸加氢还原、内酯化获得。如使用氧化铂和 2~3 atm H2(1atm=1.01325×105Pa),在室温下乙醚中还原乙酰丙酸能够得到87%的γ-戊内酯;使用兰尼镍催化剂,在120 ℃、50 atm H2条件下,γ-戊内酯的产率能够达到 94%[38]。Manzer等[39]在专利中报道了超临界CO2条件下,Ru、Pd、Pt和Rh在不同的载体如炭、二氧化钛、氧化铝和沸石上制备的负载型催化剂对乙酰丙酸加氢的效果,说明超临界CO2的存在能够促进乙酰丙酸向γ-戊内酯的转化。Poliakoff等[40]的研究也证实了这一点,并且指出超临界CO2除了能促进反应的进行,还能够在反应后起到分离产物的作用,体现了CO2的作为反应助剂的优越性。

图9 甲酸还原乙酰丙酸制备γ-戊内酯
但是需要指出的是使用非均相催化剂在 H2气氛下,由于 H2过量,不可避免地会产生少量的 2-甲基四氢呋喃,即产生了γ-戊内酯过度加氢的产物。2-甲基四氢呋喃在储存过程中会形成过氧化物,容易引发爆炸。即使产物中含有ppm级的2-甲基四氢呋喃也会存在爆炸的隐患[41]。因此,Horvath等[42]提出使用均相催化剂改进该反应。他们使用Ru(acac)3和水溶性膦配体TPPTS,在乙酰丙酸的水溶液中140 ℃、60 atm H2下得到了95%的γ-戊内酯。而当使用过量的甲酸钠作为氢源时,利用转移加氢的方法还原乙酰丙酸,γ-戊内酯的选择性较低,产率只有50%。
邓理等[43-44]利用RuCl3/PPh3催化剂实现了当量的甲酸对乙酰丙酸的加氢还原,高选择性地得到了γ-戊内酯。由于原位产生的氢气全部来自于副产物甲酸,所以该方法可以避免使用外部氢气供给。范康年等[45-46]利用负载的纳米金催化剂也能够实现当量甲酸对于乙酰丙酸的还原,且该催化剂能够催化低浓度的底物。
Manzer等[47]报道乙酰丙酸和甲酸的混合物水溶液可以通过与烯烃反应生成乙酰丙酸酯和甲酸酯(图10)。这两种酯都可以作为汽油添加剂使用,辛烷值超过100,具有很好的抗爆性。更重要的是,通过酯化这两种化合物不需要从水中进行分离就实现转化,转化后的产物不溶于水可以直接分离,大大降低了制备过程中的能耗。

图10 利用烯烃合成乙酰丙酸酯
Dumesic等[48]利用该方法也实现了乙酰丙酸丁酯和甲酸丁酯的合成与分离,并且将酯类混合物用于后续的催化转化,成功地得到95%的γ-戊内酯。
最近,美国《Science》杂志[49]报道了利用γ-戊内酯合成航空燃料的新方法(图11)。该方法利用γ-戊内酯在SiO2/Al2O3催化剂的作用下首先生成丁烯和CO2气体,再通过固体酸催化可以得到碳原子在8~16之间的低聚烯烃,产物符合航天燃料的标准,从而实现了由γ-戊内酯制备高品位燃料的过程,总产率达到60%。
通过脱羧生成的 CO2可以有效地被收集和利用,减少了生产过程中CO2的排放。更重要的是该方法所涉及的两个反应都没有使用贵金属催化剂,并且γ-戊内酯脱除氧原子没有使用任何外界 H2输入,大大提高了燃料生产过程的经济性和可持续性。但需要注意的是,研究报告中明确指出利用该方法生产航空燃料的成本取决于γ-戊内酯的价格,所以下一步工作的重点是如何设计并实现γ-戊内酯生产的新工艺,从而降低γ-戊内酯的成本。

图11 γ-戊内酯制备C8烯烃燃料
γ-戊内酯在酸性载体负载的贵金属催化剂作用下使用H2还原,能够高选择性地得到戊酸。戊酸经过氧化铈和氧化锆的混合物催化能够发生脱羧偶联反应,生成5-壬酮,再经过加氢还原可以得到汽油组分(图12)。使用Pd/NbO2催化剂,在325 ℃、3.5 MPa对50%γ-戊内酯水溶液加氢,戊酸的产率高达92%。由于戊酸具有疏水性,所以反应的产物分为油相和水相,其中油相中的碳含量占原料的95%。以油相产物为原料,在425 ℃、2 MPa、质量空速为1.1 h−1条件下,经过Zr0.5Ce0.5O2催化剂床层,5-壬酮的产率为84%[50-51]。

图12 由γ-戊内酯合成5-壬酮及烷烃
另外,Shell公司的 Lange等[52]也通过γ-戊内酯在贵金属催化剂作用下生成戊酸,再利用酯化反应将戊酸转化为戊酸酯,从而得到了更高能量密度的燃料添加剂(图13)。他们使用Pt/ZSM-5催化剂对γ-戊内酯进行加氢,戊酸的产率在 90%以上,再通过离子交换树脂催化酯化反应得到95%以上的戊酸乙酯。利用含15%戊酸乙酯的汽油,他们还成功地完成了25万千米路试,结果表明加入戊酸乙酯对于汽车引擎没有明显的影响,各项指标都达到了要求。

图13 由γ-戊内酯合成戊酸乙酯
4.3 乙酰丙酸制备化学品
利用加氢还原反应可以由乙酰丙酸得到多种化合物,包括:γ-戊内酯、2-甲基四氢呋喃、1,4-戊二醇以及戊烷。γ-戊内酯是环境友好的天然产物,在自然界存在于水果中,其毒性小于乙醇,可以用作食品添加剂。
使用均相催化剂Ru(acac)3/PBu3/NH4PF6,在8 MPa H2、200 ℃条件下对乙酰丙酸进行加氢,原料全部转化2-甲基四氢呋喃。以中间体2-甲基四氢呋喃为原料,使用催化剂Pt(acac)2/CF3SO4H组合,在150 ℃、8 MPa H2条件下反应15 h,原料全部转化为烷烃,其中丁烷和戊烷的产率分别为53%和19%。在上述条件下,不加入NH4PF6,则产物为γ-戊内酯和1,4-戊二醇,产率分别为37%和63%[42]。使用长链烷基膦配体 PnOct3和 Ru(acac)3的组合,可以以接近 100%的产率获得γ-戊内酯。而如果使用三齿膦配体Triphos和Ru(acac)3的组合,可以将1,4-戊二醇的产率提高至95%(图14)。充分体现了均相催化中配体的重要作用,也为乙酰丙酸还原产物的调控提供了有力的工具[53]。

图14 通过改变配体实现乙酰丙酸还原产物的调控
双酚A是重要的高分子单体,在工业上用来生产聚碳酸酯(PC)以及环氧树脂等高分子材料。传统的方法生产双酚A通过丙酮与两分子苯酚在酸催化下发生亲电取代反应得到。利用乙酰丙酸的羰基官能团可以替代丙酮合成双酚酸(图15),考虑到乙酰丙酸的羧基官能团,得到的高分子材料应具有较好的亲水性。利用乙酰丙酸生产双酚酸的催化剂可以是硫酸、盐酸,也可以是固体酸,如阳离子树脂和负载型的杂多酸,双酚酸的产率可以达到90%以上[54-55]。
Manzer等[56]报道了γ-戊内酯与甲醛在固体碱催化下,可以用来生产α-亚甲基-γ-戊内酯(图16),该分子作为单体可以聚合得到具有高玻璃化温度的高分子材料(PMeMBL)。以往生产α-亚甲基-γ-戊内酯通常要使用化学当量试剂,如以γ-戊内酯为原料在无水乙醚中以金属钠处理,再加入甲醛可以得到α-亚甲基-γ-戊内酯。使用当量试剂,由于价格和规模的因素通常无法实现该化合物的大规模商业化生产。Manzer等提出的方法利用 SiO2负载的乙酸钡作为催化剂,在340 ℃条件下,前15 min可以转化 60%的γ-戊内酯,得到 25%α-亚甲基-γ-戊内酯。要实现该化合物的商业化生产,提高反应的效率和催化剂寿命是亟待解决的问题。
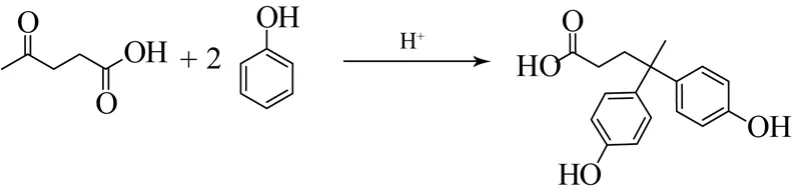
图15 利用乙酰丙酸合成水溶性双酚酸

图16 由 γ-戊内酯合成 α-亚甲基-γ-戊内酯
γ-戊内酯的另一个重要应用在于生产 3-戊烯酸甲酯,Lange等[57]利用反应蒸馏技术可以高产率地获得3-戊烯酸甲酯。使用甲醇和对甲苯磺酸(PTS)在200 ℃蒸馏γ-戊内酯,3-戊烯酸甲酯产率65%~75%(图17)。该化合物可以通过甲酰化反应、氢氰化反应和羧基化反应合成己内酯、己内酰胺和己二酸。其中合成己二酸的途径尤为引人注目,通过这种途径合成出的尼龙可以被称为“生物质基尼龙(bio-based nylon)”。

图17 由γ-戊内酯合成3-戊烯酸甲酯
5 纤维素氢解制备多元醇及其应用
5.1 纤维素加氢制备山梨糖醇
工业生产山梨糖醇一般采用兰尼镍作为催化剂,对浓的葡萄糖水溶液(来自淀粉水解)进行加氢,反应温度120~150 ℃,H2压力6~20 MPa。多种金属催化剂都能够催化葡萄糖的加氢反应,其活性顺序为 Ru > Ni > Rh > Pd[58]。尽管 Ru 催化剂的活性最高,但是其价格远远高于镍催化剂,所以工业上使用镍催化剂居多。
如果将来山梨糖醇成为生物精炼中不可或缺的重要的平台化合物,其产量势必会大大增加,基于此研究人员将目光聚集到了纤维素生产山梨糖醇的工艺上。Fukuoka等[59]率先使用纤维素作为原料,使用Pt/Al2O3作为催化剂,得到了中等产率的山梨糖醇。实验证明酸性的γ-Al2O3在贵金属的协同作用下能够产生更强的酸性,促进纤维素的水解,得到葡萄糖,再进行加氢反应得到山梨糖醇。高温热水可以原位生成大量的质子,有利于纤维素的水解。刘海超等[60]利用Ru/C催化剂在243 ℃的热水中对纤维素进行加氢,纤维素转化率85%,山梨糖醇的选择性可达35%。在反应体系中引入无机酸如磷酸或硫酸可以很好地促进纤维素水解反应的发生,从而优化制备山梨糖醇的结果。使用木材作原料,以硫酸和Ru/C作为催化剂,在160 ℃、5 MPa H2条件下反应,纤维素的转化率能够达到91%,山梨糖醇的产率达到36%(图18)[61]。
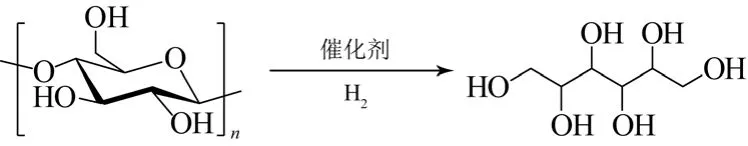
图18 纤维素催化加氢制备山梨糖醇
5.2 山梨糖醇制备燃料
与葡萄糖相比,山梨糖醇由于不含有醛基,所以在加热过程中不会形成大量的呋喃化合物,从而抑制了高温反应过程中的结焦和催化剂积炭现象,更加适合较高温度下的催化转化。山梨糖醇在燃料方面的重要应用在于其水溶液可以在温和条件下经过锡掺杂的兰尼镍催化剂重整生成H2、二氧化碳和少量烷烃,H2的选择性达到65%[62]。另外,碳水化合物还可以在自热条件下经过贵金属催化得到合成气[63]。类似的反应也可以用甘油作为原料,得到的合成气经过Ru催化剂费-托合成可以得到烷烃,上述两个反应可以在一个双固定床反应器中一步完成,大大降低了反应的能耗[64]。
Dumesic等[26]利用液相重整技术将山梨糖醇转化为轻质烷烃,从而为碳水化合物到碳氢化合物的转化搭起了桥梁(图19)。当使用Pt/ Al2O3-SiO2双功能催化剂在225 ℃重整5%的山梨糖醇水溶液可以得到轻质烷烃,产率在60%以上,且该反应连续运行6天,催化剂没有发生失活。在重整的过程中金属 Pt的作用主要是催化部分山梨糖醇分解生成H2,利用原位生成的 H2还原山梨糖醇得到烷烃。载体Al2O3-SiO2在反应中主要起到酸催化的作用,使山梨糖醇脱水形成碳碳双键,从而被H2还原。该液相重整反应的优势在于:在不使用外部H2的条件下,仅依靠反应物自身分解得到的H2实现反应物的还原。而当使用外部H2时,该反应对于己烷的选择性明显提高,产率从之前的37%提高至55%。
5.3 纤维素催化氢解直接制备乙二醇

图19 山梨糖醇液相催化重整制备烷烃

图20 纤维素催化加氢制备乙二醇
张涛等[65]在使用 W2C催化剂还原纤维素时意外地发现有大量的乙二醇生成(图20),W2C催化剂不仅能够还原纤维素,还能够选择性地切断碳碳键得到乙二醇,经过该小组的不断研究,目前由纤维素制乙二醇的最高产率可达72%。
6 结 语
本文作者综述了近期发展的、具有代表性的由纤维素催化制备生物质基平台分子的方法和途径,并分别就各种平台分子的后续转化途径进行了讨论。这其中不乏开创性的工作,如以纤维素为原料,分别经历HMF、γ-戊内酯和山梨糖醇获得烷烃的途径。相信随着更多生物质平台分子转化途径和方法的发掘,以及对于催化剂性能的改进和提高,纤维素催化制备平台化合物的研究将更加丰富和实用,这将为可再生资源替代化石资源的可持续发展提供有力的理论支持和实践指导。
[1] Werpy T,Petersen G. Top Value Added Chemicals From Biomass Volume I,Results of Screening for Potential Candidates from Sugars and Synthesis Gas[R].2004.
[2] Bozell J J,Petersen G R. Technology development for the production of biobased products from biorefinery carbohydrates-the US Department of Energy’s “Top 10” revisited[J].Green Chemistry,2010,12:539-554.
[3] Rinaldi R,Schuth F. Acid hydrolysis of cellulose as the entry point into biorefineryschemes[J].Chemsuschem,2009,2(12):1096-1107.
[4] Klemm D,Heublein B,Fink H P,et al. Cellulose:Fascinating biopolymer and sustainable raw material[J].Angewandte Chemie-International Edition,2005,44:3358-3393.
[5] Swatloski R P,Spear S K,Holbrey J D,et al. Dissolution of cellose with ionic liquids[J].Journal of the American Chemical Society,2002,124:4974-4975.
[6] Li C Z,Zhao Z K. Efficient acid-catalyzed hydrolysis of cellulose in ionic liquid[J].Advanced Synthesis & Catalysis,2007,349:1847-1850.
[7] Li C Z,Wang Q,Zhao Z K. Acid in ionic liquid:An efficient system for hydrolysis of lignocellulose[J].Green Chemistry,2008,10:177-182.
[8] Binder J B,Raines R T. Fermentable sugars by chemical hydrolysis of biomass[J].Proceedings of the National Academy of Sciences of the United States of America,2010,107:4516-4521.
[9] Feng D X,Li Z L,Zheng Y Y,et al. Separation of ionic liquid[Mmim][DMP] and glucose from enzymatic hydrolysis mixture of cellulose using alumina column chromatography[J].Appl. Microbiol.Biotechnol.,2011,91:399-405.
[10] Rinaldi R,Palkovits R,Schüth F. Depolymerization of cellulose using solid catalysts in ionic liquids[J].Angewandte Chemie-International Edition,2008,47:8047-8050.
[11] Rinaldi R,Meine N,vom Stein J,et al. Which controls the depolymerization of cellulose in ionic liquids:The solid acid catalyst or cellulose?[J].Chemsuschem,2010,3:266-276.
[12] Rinaldi R,Engel P,Buchs J,et al. An integrated catalytic approach to fermentable sugars from cellulose[J].Chemsuschem,2010,3:1151-1153.
[13] Onda A,Ochi T,Yanagisawa K. Selective hydrolysis of cellulose into glucose over solid acid catalysts[J].Green Chemistry,2008,10:1033-1037.
[14] Onda A,Ochi T,Yanagisawa K. Hydrolysis of cellulose selectively into glucose over sulfonated activated-carbon catalyst under hydrothermal conditions[J].Topics in Catalysis,2009,52:801-807.
[15] Suganuma S,Nakajima K,Kitano M,et al. Hydrolysis of cellulose by amorphous carbon bearing SO3H,COOH,and OH groups[J].Journal of the American Chemical Society,2008,130:12787-12793.
[16] Kitano M,Yamaguchi D,Suganuma S,et al. Adsorption-enhanced hydrolysis of beta-1,4-glucan on graphene-based amorphous carbon bearing SO3H,COOH,and OH Groups[J].Langmuir,2009,25:5068-5075.
[17] Yamaguchi D,Kitano M,Suganuma S,et al. Hydrolysis of cellulose by a solid acid catalyst under optimal reaction conditions[J].Journal of Physical Chemistry C,2009,113:3181-3188.
[18] Pang J F,Wang A Q,Zheng M Y,et al. Hydrolysis of cellulose into glucose over carbons sulfonated at elevated temperatures[J].Chemical Communications,2010(46):6935-6937.
[19] Lai D M,Deng L,Li J,et al. Hydrolysis of cellulose into glucose by magnetic solid acid[J].Chemsuschem,2011,4:55-58.
[20] Zhao H B,Holladay J E,Brown H,et al. Metal chlorides in ionic liquid solvents convert sugars to 5-hydroxymethylfurfural[J].Science,2007,316:1597-1600.
[21] Yong G,Zhang Y G,Ying J Y. Efficient catalytic system for the selective production of 5-hhydroxymethylfurfural from glucose and fructose[J].Angewandte Chemie-International Edition,2008,47:9345-9348.
[22] Pidko E A,Degirmenci V,van Santen R A,et al. Glucose activation by transient Cr2+dimers[J].Angewandte Chemie-International Edition,2010,49:2530-2534.
[23] Hu S Q,Zhang Z F,Song J L,et al. Efficient conversion of glucose into 5-hydroxymethylfurfural catalyzed by a common Lewis acid SnCl4in an ionic liquid[J].Green Chemistry,2009,11:1746-1749.
[24] Binder J B,Raines R T. Simple chemical transformation of lignocellulosic biomass into furans for fuels and chemicals[J].Journal of the American Chemical Society,2009,131:1979-1985.
[25] Yu S,Brown H M,Huang X W,et al. Single-step conversion of cellulose to 5-hydroxymethylfurfural(HMF),a versatile platform chemical[J].Applied Catalysis A:General,2009,361:117-122.
[26] Huber G W,Chheda J N,Barrett C J,et al. Production of liquid alkanes by aqueous-phase processing of biomass-derived carbohydrates[J].Science,2005,308:1446-1450.
[27] Huber G W,Cortright R D,Dumesic J A,Renewable alkanes by aqueous-phase reforming of biomass-derived oxygenates[J].Angewandte Chemie-International Edition,2004,43:1549-1551.
[28] Roman-Leshkov Y,Barrett C J,Liu Z Y,et al. Production of dimethylfuran for liquid fuels from biomass-derived carbohydrates[J].Nature,2007,447:982-985.
[29] Corma A,Iborra S,Velty A. Chemical routes for the transformation of biomass into chemicals[J].Chemical Reviews,2007,107:2411-2502.
[30] Schiavo V,Descotes G,Mentech J. Catalytic-hydrogenation of 5-hydroxymethylfurfural in aqueous-medium[J].Bulletin de la Societe Chimique de France,1991,128:704-711.
[31] Mascal M,Nikitin E B. Direct high-yield conversion of cellulose into biofuel[J].Angewandte Chemie-International Edition,2008,47:7924-7926.
[32] Mascal M,Nikitin E B. Towards the efficient,total glycan utilization of biomass[J].Chemsuschem,2009,2:423-426.
[33] Mascal M,Nikitin E B. High-yield conversion of plant biomass into the key value-added feedstocks 5-(hydroxymethyl) furfural,levulinic acid,and levulinic estersvia5-(chloromethyl) furfural[J].Green Chemistry,2010,12:370-373.
[34] William A F,John E C. Method for the production of levulinic:US,6054611[P]. 2000.
[35] Huber G W,Iborra S,Corma A. Synthesis of transportation fuels from biomass:Chemistry,catalysts,and engineering[J].Chemical Reviews,2006,106:4044-4098.
[36] Hu X,Li C Z. Levulinic esters from the acid-catalysed reactions of sugars and alcohols as part of a bio-refinery[J].Green Chemistry,2011,13:1676-1679.
[37] Tominaga K,Mori A,Fukushima Y,et al. Mixed-acid systems for the catalytic synthesis of methyl levulinate from cellulose[J].Green Chemistry,2011,13: 810-812.
[38] Huber G W,Iborra S,Corma A. Synthesis of transportation fuels from biomass:Chemistry,catalysts,and engineering[J].Chemical Reviews,2006,106:4044-4098.
[39] Manzer L E,Hutchenson K W. Production of 5-methyl-dihydrofuran-2-one from levulinic acid in supercritical media:US,0254384 A1[P]. 2004.
[40] Bourne R A,Stevens J G,Ke J,et al. Maximising opportunities in supercritical chemistry:The continuous conversion of levulinic acid to gamma-valerolactone in CO2[J].Chemical Communications,2007(44):4632-4634.
[41] Fabos V,Gabriella K,Hasan M,et al. Bio-oxygenates and the peroxide number:A safety issue alert[J].Energy & Environmental Science,2009,2:767-769.
[42] Mehdi H,Fabos V,Tuba R,et al. Integration of homogeneous and heterogeneous catalytic processes for a multi-step conversion of biomass:From sucrose to levulinic acid,gamma-valerolactone,1,4-pentanediol,2-methyl-tetrahydrofuran,and alkanes[J].Topics in Catalysis,2008,48:49-54.
[43] Deng L,Li J,Lai D M,et al. Catalytic conversion of biomass-derived carbohydrates into gamma-valerolactone without using an external H2supply[J].Angewandte Chemie-International Edition,2009,48:6529-6532.
[44] Deng L,Zhao Y,Li J,et al. Conversion of levulinic acid and formic acid into gamma-valerolactone over heterogeneous catalysts[J].Chemsuschem,2010,3(10):1172-1175.
[45] Du X L,He L,Zhao S,et al. Hydrogen-independent reductive transformation of carbohydrate biomass into gamma-valerolactone and pyrrolidone derivatives with supported gold catalysts[J].Angewandte Chemie-International Edition ,2011,50:7815-7819.
[46] Du X L,Bi Q Y,Liu Y M,et al. Conversion of biomass-derived levulinate and formate esters into gamma-valerolactone over supported gold catalysts[J].Chemsuschem,2011,4:1838-1843.
[47] Fagan P J,Korovessi E,Manzer L E,et al. Preparation of levulinic acid esters and from biomass and olefins:WO,03/085071A1[P].2003.
[48] Gurbuz E I,Alonso D M,Bond J Q,et al. Reactive extraction of levulinate esters and conversion to gamma-valerolactone for production of liquid fuels[J].Chemsuschem,2011,4,357-361.
[49] Bond J Q,Alonso D M,Wang D,et al. Integrated catalytic conversion ofγ-valerolactone to liquid alkenes for transportation fuels[J].Science,2010,327:1110-1114.
[50] Serrano-Ruiz J C,Wang D,Dumesic J A. Catalytic upgrading of levulinic acid to 5-nonanone[J].Green Chemistry,2010,12:574-577.
[51] Alonso D M,Bond J Q,Serrano-Ruiz J C,et al. Production of liquid hydrocarbon transportation fuels by oligomerization of biomass-derived C9alkenes[J].Green Chemistry,2010,12:992-999.
[52] Lange J P,Price R,Ayoub P M,et al. Valeric biofuels:A platform of cellulosic transportation fuels[J].AngewandteChemie-International Edition,2010,49 :4479-4483.
[53] Geilen F M A,Engendahl B,Harwardt A,et al. Selective and flexible transformation of biomass-derived platform chemicals by a multifunctional catalytic system[J].AngewandteChemie-International Edition,2010,49:5510-5514.
[54] Guo Y H,Li K X,Clark J H. The synthesis of diphenolic acid using the periodic mesoporous H3PW12O40-silica composite catalysed reaction of levulinic acid[J].Green Chemistry,2007,9:839-841.
[55] Van de Vyver S,Geboers J,Helsen S,et al. Thiol-promoted catalytic synthesis of diphenolic acid with sulfonated hyperbranched poly(arylene oxindole)s[J].Chemical Communications,2012,48:3497-3499.
[56] Manzer L E. Catalytic synthesis of alpha-methylene-gammavalerolactone:A biomass-derived acrylic monomer[J].Applied Catalysis A:General,2004,272:249-256.
[57] Lange J P,Vestering J Z,Haan R J. Towards ‘bio-based’ Nylon:conversion of gamma-valerolactone to methyl pentenoate under catalytic distillation conditions[J].Chemical Communications,2007,33:3488-3490.
[58] Wisniak J,Hershko W M,Leibowit R,et al. Hydrogenation of xylose to xylitol[J].Industrial & Engineering Chemistry Product Research and Development,1974,13:75-79.
[59] Fukuoka A,Dhepe P L. Catalytic conversion of cellulose into sugar alcohols[J].Angewandte Chemie-International Edition,2006,45:5161-5163.
[60] Luo C,Wang S A,Liu H C. Cellulose conversion into polyols catalyzed by reversibly formed acids and supported ruthenium clusters in hot water[J].Angewandte Chemie-International Edition,2007,46:7636-7639.
[61] Palkovits R,Tajvidi K,Procelewska J,et al. Hydrogenolysis of cellulose combining mineral acids and hydrogenation catalysts[J].Green Chemistry,2010,12:972-978.
[62] Huber G W,Shabaker J W,Dumesic J A. Raney Ni-Sn catalyst for H2production from biomass-derived hydrocarbons[J].Science,2003,300:2075-2077.
[63] Dauenhauer P J,Dreyer B J,Degenstein N J,et al. Millisecond reforming of solid biomass for sustainable fuels[J].Angewandte Chemie-International Edition,2007,46,5864-5867.
[64] Simonetti D A,Rass-Hansen J,Kunkes E L,et al. Coupling of glycerol processing with Fischer-Tropsch synthesis for production of liquid fuels[J].Green Chemistry,2007,9:1073-1083.
[65] Ji N,Zhang T,Zheng M Y,et al. Direct catalytic conversion of cellulose into ethylene glycol using nickel-promoted tungsten carbide catalysts[J].Angewandte Chemie-International Edition,2008,47:8510-8513.
猜你喜欢
分子催化(2022年1期)2022-11-02
化工技术与开发(2021年7期)2021-08-02
食品与发酵工业(2021年1期)2021-01-20
现代食品(2016年24期)2016-04-28
化工进展(2015年3期)2015-11-11
云南中医学院学报(2015年2期)2015-07-31
医学研究杂志(2015年5期)2015-06-10
化学反应工程与工艺(2015年1期)2015-04-16
中国塑料(2014年4期)2014-10-17
湖北农业科学(2014年13期)2014-08-28
