
气相中疏水氨基酸的单电子氧化还原性质
2010-11-06 07:01:45李伟伟侯若冰孙彦丽
物理化学学报 2010年10期
李伟伟 侯若冰 孙彦丽
(广西师范大学化学化工学院,广西桂林 541004)
气相中疏水氨基酸的单电子氧化还原性质
李伟伟 侯若冰*孙彦丽
(广西师范大学化学化工学院,广西桂林 541004)
采用密度泛函理论在B3LYP/DZP++水平上研究气相中疏水氨基酸的单电子氧化还原性质.计算表明:发生单电子氧化反应时,侧链较小的甘氨酸、丙氨酸、脯氨酸、缬氨酸、亮氨酸、异亮氨酸丢失电子的主要部位是氨基、α-碳和羧基,对应着相对较大的绝热电离能(8.52-9.15 eV);而半胱氨酸、甲硫氨酸、苯丙氨酸、酪氨酸、色氨酸因侧链丢失较多负电荷,其电离能有所降低.气相中疏水氨基酸从外界捕获的电子主要驻留在羧基或氨基的氢原子外侧以及分子的骨架上,形成具有偶极边界结构和价键结构的混合状态阴离子,绝热电子亲和势在-0.08至-0.63 eV之间.由于氨基酸的电离能较大且电子亲和势为负值,所以在气相中它们既不容易被氧化也难以被还原.
疏水氨基酸; 电离能; 电子亲和势; 氧化还原
氨基酸是蛋白质的基本组成单位,其氧化还原特征将直接影响到蛋白质的稳定性.在表征氨基酸的氧化还原性质时,电离能和电子亲和势的测定具有重要意义.1997年郭志峰等[1]采用质谱法测定了19种天然氨基酸的垂直电离能.Campbell等[2]用光电子能谱法测定了部分氨基酸的绝热电离能,但有7种氨基酸绝热电离能因实验条件限制而无法测定.在理论计算方面,Dehareng等[3]在2004年计算了甘氨酸、丙氨酸、天冬酰胺、酪氨酸和色氨酸的两种不同构象的垂直电离能,Millefiori等[4]则直接采用了文献[3]中个别氨基酸的的优势构象作为全部氨基酸的稳定构象形式计算所有氨基酸的垂直电离能,但遗憾的是其多数计算结果与实验值[1]的偏差超过0.5 eV,而最大偏差竟达2.26 eV.Kishora等[5]也计算了19种氨基酸的垂直电离能和绝热电离能,但计算值与实验值的最大偏差分别达到2.6和1.76 eV.关于氨基酸的电子亲和势,到目前为止,未见系统的研究报道.更重要的是,上述所有关于氨基酸电离能的实验和理论研究中,都缺乏电子转移过程中分子的电子结构分析,而本文则对氨基酸的绝热电离能、垂直电离能、绝热电子亲和势、垂直电子亲和势进行了更为精确的计算,同时对相关氧化还原过程中的电子结构进行了系统的分析.
所有氨基酸中,甘氨酸(Gly)、丙氨酸(Ala)、缬氨酸(Val)、亮氨酸(Leu)、异亮氨酸(Ile)、半胱氨酸(Cys)、脯氨酸(Pro)、甲硫氨酸(Met)、苯丙氨酸(Phe)、酪氨酸(Tyr)和色氨酸(Trp)等11种氨基酸均含有非极性侧链,为疏水型氨基酸,通常会形成蛋白质的疏水内核,对维持蛋白质结构稳定有重要作用.尽管天然氨基酸有20种,但限于篇幅,以及为与实验值对比,本文仅研究了气相中11种疏水氨基酸的单电子氧化还原性质.因有研究[6-7]指出气相中氨基酸的中性分子比两性离子要稳定,我们只选取具有非两性离子形式的中性氨基酸分子进行计算和分析.
1 计算方法与细节
经实验检验可靠的密度泛函理论B3LYP方法与DZP++基组[8]用于本研究的全部计算,该方法的计算结果已被广泛的实验数据[8]证实能够准确地描述分子[9]、阴离子[8]以及偶极边界态阴离子[10-12]的结构与性质,而且该方法在生物分子的反应机理[13-14]的研究也是成功的.本文中分子结构的全优化,及结构优化所得到的局部能量极小点均通过频率分析加以确认,所有计算都使用Gaussian 03程序[15]完成,体系中各原子的自然电荷布居则使用同样的计算方法根据自然键轨道(NBO)[16-20]分析得到.分子轨道和自旋密度图都使用GaussView软件绘制.
分子构象的差异对其氧化还原性质有一定影响,本文用于计算的初始结构来源于文献[21-29]报道的各个氨基酸分子的全局能量极小点.11种疏水氨基酸的构象可分为两种类型:Gly、Ala、Val、Leu、Ile和Met属构象I,而Cys、Pro、Phe、Tyr和Trp属构象II (图1).构象I、II与晶体数据库(Research Collaborator for Structural Bioinformatics Protein Data Bank)中氨基酸的构象(III)不同,为考查不同初始构象对氨基酸性质的影响,本文同时计算了以构象III为初始结构时疏水氨基酸的电离能和电子亲和势.
垂直电离能(VIP)、绝热电离能(AIP)、垂直电子亲和势(VEA)以及绝热电子亲和势(AEA)依据下列公式计算:

2 结果与讨论
计算表明,初始结构属于构象I、II的氨基酸分子的能量比初始结构为构象III的低4.53-16.11 kJ· mol-1(0.047-0.167 eV),所以,本文的后续讨论除特别说明外均针对构象I、II进行.
2.1 分子结构
优化得到的所有中性氨基酸分子都属L型,具有构象I形式的氨基酸倾向于形成O…H—N型氢键,具有构象II的氨基酸倾向于形成O—H…N型氢键,而具有构象III的氨基酸则形成(H)O…H—N型氢键(图1).
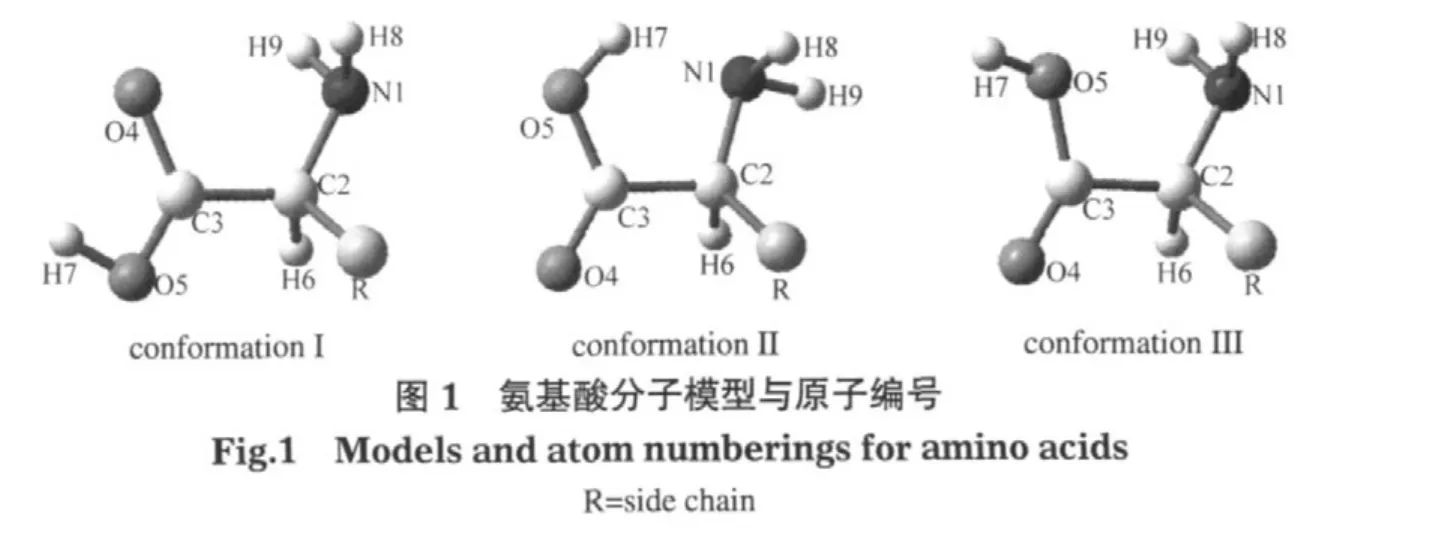
氨基酸分子丢失一个电子后,所产生的多数阳离子自由基(Gly+、Ala+、Val+、Ile+、Leu+和Pro+)中C2—N1键长较中性分子缩短6-8 pm.出现这种变化的原因在于中性氨基酸分子的最高占据轨道(HOMO)在C2—N1键附近具有π反键轨道特征,反键电子的丢失加强了C2、N1间的成键作用而使键长缩短.此外,Gly+、Ala+、Leu+和Pro+的C2—C3键长比中性分子的伸长8-13 pm,Val+、Ile+的C2与侧链R的键连C原子间的共价键C2—C(R)伸长了17 pm,究其原因是这些分子的HOMO在这两个成键位置都具有σ成键轨道特征,成键电子的丢失明显削弱了成键作用使键长伸长,这种共价键的削弱有可能造成氨基酸分子脱羧或解体.
氨基酸捕获一个电子后,形成的阴离子自由基与中性分子相比较,两体系化学键长变化都小于3 pm,键角变化小于5°.这与多数氨基酸阴离子表现为偶极边界结构和共价结构的混合结构状态有关,即阴离子捕获的外来电子只有一部分进入了氨基酸分子骨架的轨道中,与此同时,还有一部分电子则分布在分子外侧的某个部位.外来电子的这种分布方式对中性氨基酸分子中电子分布的影响不是很大,所以,形成的阴离子与中性分子相比,结构变化不明显.
2.2 分子轨道分析
图2中只绘出了部分具有代表性的中性分子的HOMO,最低空轨道(LUMO),以及阴离子自由基的HOMO(即其单电子占据轨道SOMO).
中性分子中,Val(图2)、Gly、Ala、Pro、Ile和Leu的HOMO主要分布在分子的羧基、氨基、C2及侧链第一个C原子的周围,由此可以推测,这六种氨基酸氧化丢失电子的部位应主要集中在这些原子附近.NBO电荷计算表明,氧化过程中Gly、Ala、Pro和Leu的N1和O4原子丢失电子0.53e-0.61e,C3原子丢失0.02e-0.05e;而Val、Ile的N1原子分别丢失0.41e、0.40e,侧链第一个C原子都约丢失0.11e,这些电荷转移过程与阳离子中C2—C3或C2—C(R)键明显伸长有一定关系.Phe的HOMO(图2)分布在O4和侧链芳环上,氧化过程中Phe的O4和芳环上共丢失电子0.78e.由于Cys(图2)、Met的HOMO主要分布在侧链的S原子上,Trp(图2)和Tyr的HOMO则是主要分布在侧链的共轭环上,因此,这四种氨基酸的氧化反应主要发生在侧链上,其中Cys和Met的S原子分别丢失电子0.56e、0.51e,Trp和Tyr的共轭环丢失电子0.88e、0.73e,而这与Vorsa等[30]的实验结论是一致的.

侧链中无芳环的 Val、Gly、Ala、Ile、Leu、Cys、Pro和Met,其LUMO都分布在羧基上,Trp和Tyr的LUMO则分布在侧链的芳环上,而Phe的LUMO在羧基和侧链苯环上都有分布.如前一节所述,在中性氨基酸分子的单电子还原反应中,外来电子并没有完全进入中性分子的LUMO,所形成阴离子的SOMO在分子的外侧和分子骨架上都有分布,形成兼具偶极边界阴离子和价键结构阴离子的混合结构.具有构象 I的 Val-、Gly-、Ala-、Leu-、Ile-的SOMO主要分布在阴离子羧基H原子的外侧以及其它多个原子周围,Met-的SOMO则主要分布在羧基上,少量分布在H6原子的外侧;具有构象II的Cys-和Pro-的SOMO主要分布在氨基H原子,以及C2、C3等多个原子上,Trp-、Tyr-和Phe-的SOMO在氨基H原子外侧、羧基和芳环的多个原子周围都有分布,而且,Phe-的SOMO在整个分子中离域程度最大.目前,已有实验[31-32]表明Gly-和Ala-确实具有一定的偶极边界离子特征,这种电荷分布形式的重要性在于氨基酸阴离子易于把多余的负电荷释放到外界环境(如溶剂水)中,从而减少外来电子对蛋白质的损伤.
2.3 自旋密度
自旋密度可以反映自由基中单电子的分布,根据自由基中的自旋密度分布可以推测自由基的氧化还原性特征.图3为部分代表性的氨基酸离子自由基的自旋密度图.11种疏水氨基酸阳离子自由基的自旋密度分布在形态上与其中性分子的HOMO基本一致,表明中性分子的电离过程确实发生在其HOMO上,电离形成的阳离子自由基的单电子多驻留在电负性较大的N、O原子以及S原子和侧链芳环上.对于侧链无芳环的阳离子(如Val+,Cys+),其自旋密度分布使得阳离子具有从外界夺取电子还原为中性分子的倾向.事实上,中性分子较大的VIP值(表1)也是阳离子这种氧化性倾向的一种反映.
氨基酸阴离子自由基的自旋密度分布都与其SOMO基本一致,除在分子骨架中有部分自旋密度分布外,图3显示多数氨基酸阴离子的单电子也明显地驻留在电负性较小的H原子附近,这种自旋密度分布形式不利于阴离子对其电子的控制,易失去电子而呈还原性.值得注意的是Phe-的自旋密度在侧链的芳环上存在非常明显的离域分布,这种分布形态显然有利于阴离子中电荷的分散,进而增强Phe-的稳定性.
2.4 电离能和电子亲和势
表1列出了计算的电离能与电子亲和势,所有疏水氨基酸的VIP都大于AIP,VEA的绝对值都比AEA的大一些,这是合理的,因为氨基酸分子在失去或得到一个电子后,最初的阳、阴离子随后的结构松弛将使体系能量下降,形成相对稳定的结构状态.
多数氨基酸VIP的计算结果与实验值[1]比较接近,VIP计算值与实验值的平均偏差为0.34 eV.尽管Ala、Cys和Trp的VIP计算值与实验值偏差较大(0.48-0.66 eV),但我们的计算精度仍优于文献的结果[4-5].这些文献报道的VIP计算值与实验值平均偏差远大于本文的0.34 eV,分别达到了0.49和0.96 eV.而他们对Ala、Cys和Trp的计算结果与实验值的偏差范围则分别为0.82-1.18 eV和0.83-2.05 eV,这表明基于B3LYP/DZP++方法计算的VIP值更可靠.

表1 气态中疏水氨基酸的电离能和电子亲和势Table 1 Ionization potentials and electron affinities in gas phase
本文对疏水氨基酸AIP的计算相当精确,除Cys的计算值比实验值[2]高出0.56 eV外,整体计算值与实验值的平均偏差仅为0.24 eV.文献[5]也计算了疏水氨基酸的AIP,但其计算值与实验值的平均偏差却达到了0.64 eV,其Cys的计算结果比实验值也高出0.68 eV.
所有疏水氨基酸电子亲和势的数值都为负值,这说明中性分子的能量要比其阴离子能量稍低,气相中阴离子不能稳定存在,这显然与所形成的阴离子兼具偶极边界结构态和价键结构态,负电荷不能充分地分散在整个阴离子中有关.
仔细观察表1还可以发现,氨基酸电离能的大小与侧链有密切关系,侧链上C原子数越多,其电离能越小.而Cys,Met侧链上S原子使它们的电离能比侧链中所含有碳原子数相同的氨基酸的要小,其中Met的电离能甚至比Phe的还小,这一现象与氨基酸的结构及其丢失电子的部位密切相关.正如前面分子轨道的分析所指出的,含脂肪烃基侧链的氨基酸分子丢失电子的部位是氨基和羧基,但含S的氨基酸的电离同时也发生在S原子附近,而S的电负性与C相当,却比N和O要小得多,因而对电子的束缚能力较弱,故含S的氨基酸Cys和Met易失去电子.
具有同一构象形式的疏水氨基酸电子亲和势的数值也会随着侧链碳原子数的增多而减小.究其原因是因为氨基酸侧链具有分散电荷的作用,随着体系的增大,其侧链容纳和分散负电荷的能力增强,因而可以形成相对稳定些的阴离子.
2.5 氨基酸分子的初始构象对垂直电离能的影响
仔细分析表1的电离能数据,可观察到一个有趣的现象——使用全局能量极小点分子结构(构象I、II)作初始结构计算出的VIP值(数据Theor1)均大于初始结构源于蛋白质数据库(构象III)的计算值(数据Theor2).这表明具有不同构象的氨基酸分子垂直电离时所需的能量是不同的,所有11个疏水氨基酸分子因初始构象差异而导致的垂直电离能的差别在0.04-0.55 eV范围内,这是具有全局能量极小点构象的氨基酸分子垂直电离时环境需要多付出的能量.当然,发生垂直电离时环境提供的能量总是过量的,因此,无论是哪种构象的氨基酸分子,其电离过程都能进行.
另一方面,如果将气相氨基酸视为服从Boltzmann分布、非简并、可分辨的分子体系,那么,就能容易地估算出具有不同能量、不同构象的氨基酸分子发生垂直电离的分子百分数,进而可利用该百分数做权重估算出各氨基酸统计平均的VIP值.为简便起见,我们假定氨基酸气体中只存在I、III或II、III两种构象态,这里以两种构象态能量差最大的Leu(16.11 kJ·mol-1)和最小的Ala(4.53 kJ·mol-1)为例进行计算.根据Boltzmann分布关系(式中kB、T分别为Boltzmann常数和绝对温度):

可估算出在文献[1]的实验温度260℃时,具有能量ε1(构象I或II)、ε2(构象III)的两种分子的分子数n1、n2之比.Leu和Ala的该比值分别为37.94和2.782,也就是说,两种氨基酸的低能量构象的分子百分数分别为97.43%和73.56%,以此为权重计算得到的统计平均VIP值分别为9.31、9.54 eV.这两个计算值与表1中的数据对比可知,由于具有高能量的构象III的分子百分数较小,而且,单独利用构象III的结构计算出的VIP值又偏小,所以,统计平均的VIP值只是比单独利用构象I或II计算出的值略小.我们将全部氨基酸的VIP值按这种方式计算出来后进行统计,发现统计平均的VIP值与实验值的平均偏差比全部使用构象I、II结构的VIP计算值的平均偏差0.34 eV稍小,但如果全部使用构象III的结构计算VIP值,则对应的平均偏差较大(0.42 eV).由此可见,全面考虑各种可能的初始构象计算出的VIP值更为可靠.
2.6 蛋白质-DNA复合物单电子损伤的分析
任何生物体的生长、发育过程都离不开蛋白质-DNA间的相互作用.在正常条件下,只有当蛋白质与DNA间的准确识别过程顺利完成后,才能有效地形成蛋白质-DNA复合物,随后进行DNA的复制,新蛋白质的合成等生命过程.但在这些复杂的过程中,只要在蛋白质-DNA间的相互作用的环节上发生一个微小的氧化还原变化,就可能出现蛋白质或DNA损伤,这类氧化还原损伤如果不能得到有效的修复,便会对生命体的安全产生严重威胁.
比较气相中11种疏水氨基酸与DNA中4种单核苷酸的氧化还原性质有助于我们分析和推测蛋白质-DNA复合物发生氧化还原损伤时的可能对象.使用相同的理论方法,我们计算了DNA中4种单核苷酸的氧化还原性质.2′-腺嘌呤脱氧核苷-5′-磷酸、2′-鸟嘌呤脱氧核苷-5′-磷酸[33]、2′-胸腺嘧啶脱氧核苷-5′-磷酸、2′-胞嘧啶脱氧核苷-5′-磷酸的AIP和AEA分别为7.49、7.13、7.84、7.71 eV和0.24、0.17、0.38、0.23 eV.显然,如果蛋白质-DNA复合物中不同的核苷酸与不同的疏水氨基酸相互作用时遭遇到氧化还原的威胁,所发生的具体损伤结果是不同的.由于氨基酸的AEA均为负值,而4种脱氧单核苷酸的AEA均为正值,所以,外界因素所带来的电子将只会引起DNA损伤.另一方面,由于4种脱氧单核苷酸的AIP几乎都比疏水氨基酸的小(Trp除外,为7.2 eV),所以当外来的氧化性微粒出现在蛋白质-DNA复合物附近时,也会导致DNA出现损伤,只有当蛋白质-DNA复合物附近恰好存在的氨基酸残基是Trp时,损伤的对象才可能是蛋白质.尽管这种分析只是建立在气相计算结果的基础之上,没有考虑到水的存在,但在蛋白质-DNA复合物的疏水相互作用区域里,这种推测是合理的.因为在疏水区域里,完全溶剂化是不存在的,至多只是微溶剂化(水分子极少)而已,因此,这种现象值得实验上的进一步研究.
3 结 论
使用可靠的B3LYP/DZP++理论方法探讨了气相中11种疏水氨基酸的单电子氧化还原性质.理论计算表明:
(1)氨基酸分子发生单电子氧化反应时,侧链结构不同,丢失电子的部位有差异,Gly、Ala、Pro、Val、Ile和Leu主要在H2N—C2—COOH部位丢失电子, Phe的羰基O和侧链部位都会丢失电子,而Cys、Met、Tyr和Trp的电子丢失则主要发生在侧链.
(2)轨道分析和自旋密度分析一致表明,气相中疏水氨基酸从外界捕获一个电子发生单电子还原反应后,形成了兼具偶极边界结构与价键结构的混合状态阴离子,单电子主要驻留在电负性小的H原子外侧以及分子骨架的部分原子周围.
(3)多数疏水氨基酸电离能的计算值与实验值相当接近,电离能数值较大,电子亲和势数值为较小的负值,表明氨基酸在生物体内既不容易被氧化也难以被还原.由于DNA中4种单核苷酸的电离能相对小而电子亲和势却为正值,因而当蛋白质-DNA复合物的疏水相互作用区域遭遇到氧化性或还原性微粒时,容易造成损伤的是DNA而不是蛋白质.
1 Guo,Z.F.;An,Q.R.Journal of Hebei University:Natural Science Edition,1997,17:73 [郭志峰,安秋荣.河北大学学报(自然科学版),1997,17:73]
2 Campbell,S.;Beauchamp,J.L.;Rempe,M.;Lichtenberger,D.L. Int.J.Mass Spectrom.Ion Processes,1992,117:83
3 Dehareng,D.;Dive,G.Int.J.Mol.Sci.,2004,5:301
4 Millefiori,S.;Alparone,A.;Millefiori,A.;Vanella,A.Biophys. Chem.,2008,132:139
5 Kishora,S.;Dhayalb,S.;Mathurc,M.;Ramaniah,L.M.Mol. Phys.,2008,106:2289
6 Wright,L.R.;Borkman,R.F.J.Am.Chem.Soc.,1980,102:6207
7 Rai,A.K.;Song,C.;Lin,Z.J.Spectrochim.Acta A,2009,73: 865
8 Rienstra-Kiracofe,J.C.;Tschumper,G.S.;Schaefer,H.F.;Nandi, S.;Ellison,G.B.Chem.Rev.,2002,102:231
9 Hou,R.B.;Gu,J.D.;Xie,Y.M.;Yi,X.H.;Schaefer,H.F. J.Phys.Chem.B,2005,109:22053
10 Richardson,N.A.;Gu,J.;Wang,S.;Xie,Y.;Schaefer,H.F.J.Am. Chem.Soc.,2004,126:4404
11 Gu,J.;Xie,Y.;Schaefer,H.F.Nucleic Acids Res.,2007,35:5165
12 Gu,J.;Xie,Y.;Schaefer,H.F.J.Phys.Chem.B,2010,114:1221
13 Bao,X.G.;Wang,J.;Gu,J.D.;Leszczynski,J.Proc.Natl.Acad. Sci.U.S.A.,2006,103:5658
14 Hou,R.B.;Li,W.W.;Shen,X.C.Acta Phys.-Chim.Sin.,2008, 24:269 [侯若冰,李伟伟,沈星灿.物理化学学报,2008,24: 269]
15 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.,Gaussian 03. Revision B.03.Pittsburgh,PA:Gaussian Inc.,2003
16 Reed,A.E.;Weinstock,R.B.;Weinhold,F.J.Chem.Phys.,1985, 83:735
17 Reed,A.E.;Weinhold,F.J.Chem.Phys.,1985,83:1736
18 Reed,A.E.;Curtiss,L.A.;Weinhold,F.Chem.Rev.,1988,88: 899
19 Reed,A.E.;Schleyer,P.v.R.J.Am.Chem.Soc.,1990,112:1434
20 Glendening,E.D.;Reed,A.E.;Carpenter,J.E.;Weinhold,F.NBO Version 3.1
21 Pacios,L.F.;Gomez,P.C.J.Mol.Struct.-Theochem,2001,544: 237
22 Lu,J.F.;Zhu,S.L.;Zhou,Z.Y.;Wu,Q.Y.;Zhao,G.Pol.J. Chem.,2006,80:471
23 Wilke,J.J.;Lind,M.C.;Schaefer III,H.F.;Csaszar,A.G.;Allen, W.D.J.Chem.Theory Comput.,2009,5:1511
24 Czinki,E.;Csazar,A.G.Chem.Eur.J.,2003,9(4):1008
25 Dokmaisrijan,S.;Lee,V.S.;Nimmanpipug,P.J.Mol.Struct.-Theochem,2010,953:28
26 Armentrout,P.B.;Gabriel,A.;Moision,R.M.Int.J.Mass Spectrom.,2009,283:56
27 Huang,Z.J.;Yu,W.B.;Lin,Z.J.J.Mol.Struct.-Theochem,2006, 758:195
28 Zhang,M.L.;Huang,Z.J.;Lin,Z.J.J.Chem.Phys.,2005,122: 134313
29 Huang,Z.J.;Lin,Z.J.J.Phys.Chem.A,2005,109:2656
30 Vorsa,V.;Kono,T.;Willey,K.F.;Winograd,N.J.Phys.Chem.B, 1999,103:7889
31 Diken,E.G.;Hammer,N.I.;Johnson M.A.J.Chem.Phys.,2004, 120:9899
32 Abouaf,R.Chem.Phys.Lett.,2008,451:25
33 Hou,R.B.;Li,W.W.Journal of Guangxi Normal University: Natural Science Edition,2009,27:78 [侯若冰,李伟伟.广西师范大学学报(自然科学版),2009,27:78]
Characteristics of One Electron Redox Behavior of Hydrophobic Amino Acids in Gas Phase
LI Wei-Wei HOU Ruo-Bing*SUN Yan-Li
(College of Chemistry and Chemical Engineering,Guangxi Normal University,Guilin 541004,Guangxi Province,P.R.China)
Characteristics of the one electron redox behavior of hydrophobic amino acids in gas phase were calculated with density functional theory at the B3LYP/DZP++level.For glycine,alanine,proline,valine,leucine,and isoleucine with small side chains,the computational results indicate that the negative charges are removed from the atoms of their amino,α-carbon,and carboxy moieties in one electron oxidation reactions.This yields large adiabatic ionization potentials(AIP)of 8.52-9.15 eV.The AIPs of cysteine,methionine,phenylalanine,tyrosine,and tryptophan decrease because of the larger amount of negative charge removed from the atoms in their side chains.The attachment of one electron to the molecules of hydrophobic amino acids leads to anions in which the extra electron is bound to the H atoms of the carboxyl or amino groups and to their valence orbitals,reflecting the double nature of the dipole-bound state and the valence state.The electron affinities(EA)for the amino acids are small and negative ranging from-0.08 to-0.63 eV.The molecules of the hydrophobic amino acids are oxidized or reduced with difficulty in gas phase because of their high VIPs and negative EAs.
Hydrophobic amino acid; Ionization potential; Electron affinitiy; Redox
O641
Received:May 26,2010;Revised:June 30,2010;Published on Web:August 17,2010.
*Corresponding author.Email:rbhou@163.com;Tel:+86-773-5846279.
The project was supported by the Innovation Project of Guangxi Graduate Education,China(2009106020703M48).
广西研究生创新计划项目(2009106020703M48)资助
ⒸEditorial office of Acta Physico-Chimica Sinica
猜你喜欢
——以物质结构与性质模块“元素周期律”教学为例
化学教与学(2023年3期)2023-02-09 08:33:22
山东农业大学学报(自然科学版)(2021年3期)2021-07-29 03:07:02
世界农药(2019年2期)2019-07-13 05:55:12
读与写·上旬刊(2018年10期)2018-11-27 20:03:08
粘接(2017年4期)2017-04-25 08:37:20
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
应用化工(2014年7期)2014-08-09 09:20:23
化学教与学(2014年6期)2014-07-03 10:04:09
无机化学学报(2014年5期)2014-02-28 17:31:40
