
酯加氢反应中影响羧基活化的因素
2010-11-30 10:50:08郑小娟周娅芬付海燕李贤均李瑞祥
物理化学学报 2010年10期
郑小娟 周娅芬 付海燕 陈 华 李贤均 李瑞祥
(四川大学化学学院,教育部绿色化学与技术重点实验室,成都 610064)
酯加氢反应中影响羧基活化的因素
郑小娟 周娅芬 付海燕 陈 华 李贤均 李瑞祥*
(四川大学化学学院,教育部绿色化学与技术重点实验室,成都 610064)
通过浸渍法制备4%Ru-9%La/γ-Al2O3催化剂,采用X射线衍射(XRD),X射线光电子能谱(XPS)和透射电子显微镜(TEM)对其结构进行表征.将该催化剂用于丙酸甲酯的加氢反应,分别考察了溶剂、无机盐添加剂、底物空间因素及电子因素对酯加氢反应的影响.发现以水为溶剂以及添加Co(NO3)2对丙酸甲酯加氢都显示出明显的促进作用,底物的转化率及丙醇的选择性随之增加.这是由于水及适量Co2+的引入能极化底物分子的C=O键,有利于活化氢对羧基碳原子的进攻,改善催化剂的性能.此外,底物分子中吸电子基团能提高羧基碳原子的正电性,也有利于加氢反应的进行;而增大底物分子空间位阻,不利于底物在催化剂上的吸附,催化反应速率下降.
催化加氢;丙酸甲酯;活化;钌; 镧
醇是化学和医药工业中重要的中间体,可以由含羰基的化合物如醛、酮、羧酸及其酯或酐加氢得到.然而由于酯基中C=O双键有较大的空间位阻和弱的极性使得羧酸酯还原成醇十分困难[1].工业上通常用铜锌、铜铬等复合氧化物催化羧酸及其酯加氢[2-3],加氢需要在高温(250-300℃)和高压(25-35 MPa)下进行.后来,人们对钌、铑等贵金属作为主要活性组分的催化剂用于酯加氢进行了研究.但单金属催化剂对羧酸及酯的加氢活性和选择性往往不高[4],为此,人们通过引入一种或多种金属来调变单金属催化剂的性能,对酯加氢取得了较高的活性和醇选择性.例如用Rh-Sn/SiO2,Ni-Sn/SiO2和Ru-Sn/ SiO2双金属催化剂成功对乙酸酯进行了气相加氢制备乙醇[5].双金属催化剂Ru-Sn/SiO2[5]、Ru-Sn/TiO2[6-7]、Ru-Sn-B/Al2O3[8]、Co-Sn/Al2O3[9]、Ru-Sn/Al2O3[10-11]、Rh-Sn/Al2O3[12]被广泛地用于直链羧酸及酯的加氢,取得了不错的效果.文献报道[11,13]负载Ru-Sn催化剂催化酯加氢反应中,SnOx物种(Sn2+或Sn4+)作为Lewis酸与羧基氧结合极化了C=O键,从而改善了酯加氢的性能.本课题组[14]用Ru-Pt/AlO(OH)对丙酸甲酯加氢发现,溶剂H2O和载体AlO(OH)的表面羟基与底物的羧基氧能形成氢键,同样可以极化C=O键,并且加速了反应的进行.本文在此基础上,以丙酸甲酯为模型化合物,进一步研究在溶液中添加无机盐,以及底物空间和电子因素对羧基活化的影响.
1 实验部分
1.1 试 剂
γ-Al2O3(AR,100-160目,山西日化所,500℃焙烧4 h);RuCl3·nH2O(AR,昆明贵金属研究所);LaCl3· 7H2O、SnCl4、SnCl2、ZnSO4、Co(NO3)2、Ca(NO3)2、FeSO4、FeCl3均为分析纯;乙醇、正丙醇、异丙醇、正丁醇(AR,成都长联化工试剂有限公司);丙酸甲酯、丙酸乙酯、丙酸、乙酸乙酯、三氟乙醇、三氟乙酸乙酯等均为分析纯.丙酸正丙酯、丙酸异丙酯、丙酸正丁酯、丙酸三氟乙酯自制;氢气纯度大于99.99%(四川天一科技股份有限公司).
1.2 催化剂的制备及表征
4%Ru-9%La/γ-Al2O3制备.将一定量的LaCl3· 7H2O水溶液加入到γ-Al2O3中,室温搅拌过夜后旋干,120℃干燥24 h,350℃煅烧3 h,得催化剂前驱体La2O3/γ-Al2O3.将一定量的RuCl3溶液加入到悬浮了La2O3/γ-Al2O3的水溶液中,室温下搅拌过夜后旋干,转入高压釜中以水为溶剂,在180℃和5.0 MPa条件下,加氢还原3 h.固体物质经过滤,去离子水洗涤至无氯离子,60℃真空干燥8 h,即得催化剂.洗涤后的滤液收集用于测定金属担载量,经电感耦合等离子体光谱(ICP)测得金属担载的Ru的质量分数为4%,La2O3/γ-Al2O3质量比为9%.
催化剂的X射线光电子能谱(XPS)在英国Kratos公司的XSAM 800型X射线光电子能谱仪上测得,激发源为Al Kα(1215 eV),电流15 mA,电压12 kV,用C 1s结合能284.8 eV定标.X射线衍射(XRD)采用日本理学D/max-rA旋转阳极X射线衍射仪,Cu Kα射线,λ=0.154 nm,管电压50 kV,管电流40 mA,扫描区间为20°-110°.催化剂的形貌与金属粒子大小通过日本JEOL公司的JEM-1200EX型透射电子显微镜测定,加速电压为100 kV.金属担载量用ICP(美国热电元素公司IRIS Advantage型光谱仪)测定.
1.3 催化加氢反应
底物的催化加氢反应在60 mL带磁力搅拌装置的不锈钢高压釜中进行.将一定量催化剂、反应底物和溶剂加入高压釜中,闭釜后用高纯氢置换反应釜5次,充氢气至所需压力,升温至设定温度开始搅拌并计时(搅拌速度为1500 r·min-1).反应结束后,取出样品,过滤除去催化剂,加氢产物在安捷伦GC-6890气相色谱仪上分析,SE-30石英毛细管柱(30 m×0.25 mm×0.25 μm,美国Supelco公司).采用峰面积归一化法计算含量.
2 结果与讨论
2.1 催化剂的表征
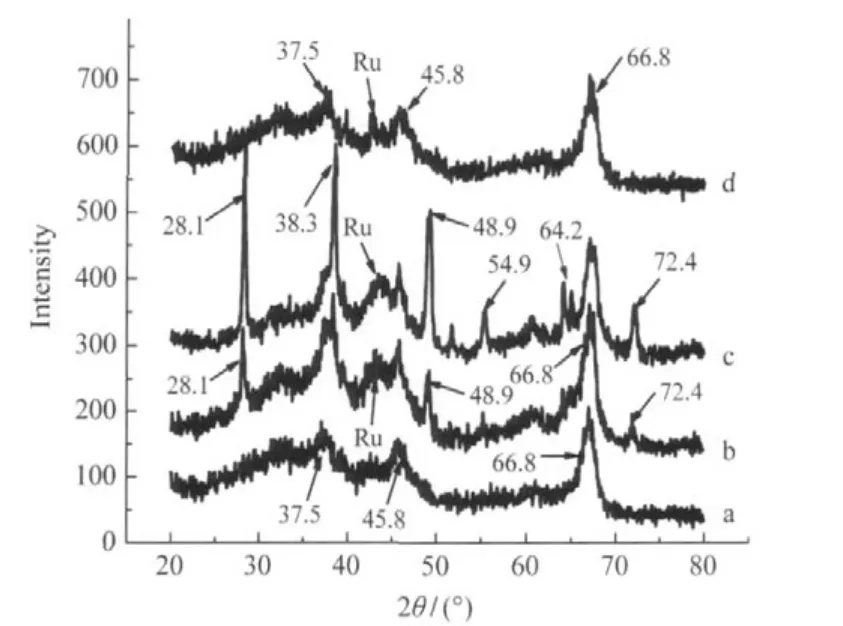
图1 4%Ru-9%La/γ-Al2O3催化剂的XRD图Fig.1 XRD patterns of 4%Ru-9%La/γ-Al2O3catalyst (a)not reduced,(b)reduced with H2in water for 3 h,(c)afterhydrogenation in water for 8 h;(d)reduced with H2in ethanol for 3 h; (b-d)temperature:180℃,H2pressure:5.0 MPa
催化剂4%Ru-9%La/γ-Al2O3样品a-d经不同处理:未经还原(样品a);以水做溶剂、180℃、5.0 MPa H2下还原3 h(样品b);还原、并用水做溶剂、180℃、5.0 MPa H2下反应8 h(样品c)以及以乙醇做溶剂、180℃、5.0 MPa H2下还原3 h(样品d)后的XRD谱见图1.图1a中2θ为37.5°、45.8°、66.8°的弥散峰属于γ-Al2O3的衍射峰(JCPDS No.29-63),可能由于金属担载量低分散好的缘故,均没有观察到Ru、RuO、Ru2O的衍射峰[15].图1b和图1c中2θ为28.1°、38.3°、48.9°、54.9°、64.2°及72.4°的峰为薄水铝石的衍射峰(γ-AlO(OH),JCPDS No.21-1307).表明催化剂在水热条件下还原以及水作溶剂条件下反应后,γ-Al2O3转变成为AlO(OH)[16].而且反应8 h后的催化剂,这些峰变得更强更尖锐,说明AlO(OH)在催化反应过程中进一步生长变大.图1d中只发现2θ为37.5°、45.8°、66.8°的γ-Al2O3衍射峰,并没发现薄水铝石的衍射峰,说明催化剂在乙醇中还原时,γ-Al2O3没有转变成γ-AlO(OH).而且还原和使用后催化剂的XRD图中(图1(b,c,d)),在2θ=43°处出现一个弱而弥散的Ru0衍射峰(JCPDS No.6-0663),表明金属颗粒在加氢过程中发生了团聚.
图2为催化剂未经还原(图2a),以水为溶剂、在180℃、5.0 MPa H2条件下还原3 h(图2b),及以乙醇为溶剂、在180℃、5.0 MPa H2条件下还原3 h(图2c)后的TEM图.从图2b中可以看到大的薄片状薄水铝石载体 ,同时也有少量未完全转化的γ-Al2O3,而图2(a,c)中载体均为纤维状的γ-Al2O3,这与XRD谱图结果一致.另外,从几个图上都可看到,金属粒子约为2-5 nm,且均匀分布于载体上.
图3是该催化剂反应后(水做溶剂,180℃、5.0 MPa H2,反应时添加Co(NO3)2)的XPS图谱,从图3a上可见Ru 3d5/2的结合能位于280.13 eV,与Ru0的结合能十分接近,证明催化剂中的Ru以零价态形式存在[18].而La 3d5/2(图3b)的电子结合能834.89 eV,与文献中La2O3(834.92 eV,La3+)的值[19]几乎一样,说明反应后镧的氧化态没有发生变化.Co的电子结合能为781.6 eV(图3c),表明Co在反应后氧化态也没有发生变化[20].
2.2 溶剂对酯加氢反应的影响

图2 不同催化剂的TEM图Fig.2 TEM images of different catalysts (a)not reduced;(b)reduced with H2in water for 3 h;(c)reduced with H2in ethanol for 3 h (b,c)temperature:180℃,H2pressure:5.0 MPa

图3 4%Ru-9%La/γ-Al2O3催化剂在水中反应8 h后的XPS图谱Fig.3 XPS spectra of the 4%Ru-9%La/γ-Al2O3catalyst after hydrogenation in water for 8 h temperature:180℃,H2pressure:5.0 MPa
溶剂对丙酸甲酯加氢反应的影响的结果列于表1.当催化剂在水中以H2还原,催化反应以水作溶剂时(entry 1),反应表现出高的活性和醇选择性.而以乙醇或乙醇-水混合物作溶剂时(entry 2和entry 3),不仅转化率比水作溶剂时低,而且生成较多的酯交换产物.说明水能抑制酯交换反应的发生[21].由entry 1和entry 4结果可以看出,以水作反应溶剂时,在水中还原的催化剂又比在乙醇中还原的催化剂活性更高.根据XRD、TEM表征结果,乙醇中还原的催化剂,载体在还原过程中结构没有发生变化,仍为γ-Al2O3;而在水中还原的催化剂,γ-Al2O3转变成了薄水铝石.我们以前研究Ru-Pt/AlO(OH)对丙酸甲酯加氢反应发现,载体丰富的表面羟基和溶剂水的协同作用能促进反应活性和选择性的提高.一方面, AlO(OH)的表面羟基与底物的羧基氧形成氢键,极化了C=O键,使Ru上活化的氢对C=O键上的C原子进攻更加容易[14];另一方面,水分子中碱性氧原子易于与配位H2结合,促进其异裂得到Ru—H和质子化的水[22-23],然后Ru上H-进攻羧基上缺电子的C原子形成中间产物醛[13],醛迅速加氢得到产物醇.而本文制备的催化剂在水热条件下转化为Ru-La/AlO(OH),表现出相似的性质.
2.3 无机盐添加剂对酯加氢反应的影响
通过溶剂的选择研究发现,底物与溶剂间形成氢键可以活化羧基,使加氢反应更易发生[14].而金属离子对—NO2的活化作用也曾有文献报道 ,认为金属离子作为路易斯酸能与—NO2中作为路易斯碱的氧原子作用,从而活化—NO2中的N=O键,在加氢条件下实现了—NO2快速还原为—NH2.在文献报道的Ru-Sn双金属催化酯加氢机理中[11,13],作者同样认为SnOx物种(Sn2+或Sn4+)作为路易斯酸与酯中羧基氧结合极化了C=O键,从而利于活化氢对C=O键上碳原子的进攻.因此,我们为了更清楚地理解羧基活化的原理,将金属离子直接添加到反应溶液中,考察其对酯加氢的影响.
表2列出了各种无机盐添加剂对丙酸甲酯加氢反应的影响.数据显示,添加适量的Co2+能明显提高底物的转化率,醇的选择性也保持不变.当添加的Co(NO3)2溶液使得Co/Ru摩尔比为4时,丙酸甲酯的转化率得到了最大程度的提高,从82.2%上升到了96.8%.继续增大Co/Ru比,底物的转化率和醇选择性都降低.然而其它的添加剂对催化活性没有明显改善,有些反而还使活性降低.
为了进一步探讨Co2+在反应中是如何对反应起促进作用时,我们进一步考察了Co2+的添加方式对反应的影响,结果见表3所示.方式I是将RuCl3和Co(NO3)2溶液共浸渍到催化剂前体La/γ-Al2O3上,然后300℃煅烧4 h制得;方式Ia是与I同样方式负载但不煅烧.方式II是将制成的Ru-La/γ-Al2O3催化剂再浸渍Co(NO3)2溶液,最后300℃煅烧4 h制得;IIa是与II同样方式负载但不煅烧.方式III是用已制备好的Ru-La/γ-Al2O3作催化剂,将Co(NO3)2直接加入反应的溶液中.

表1 溶剂对丙酸甲酯加氢反应的影响Table 1 Effect of solvent on the hydrogenation of methyl propionate
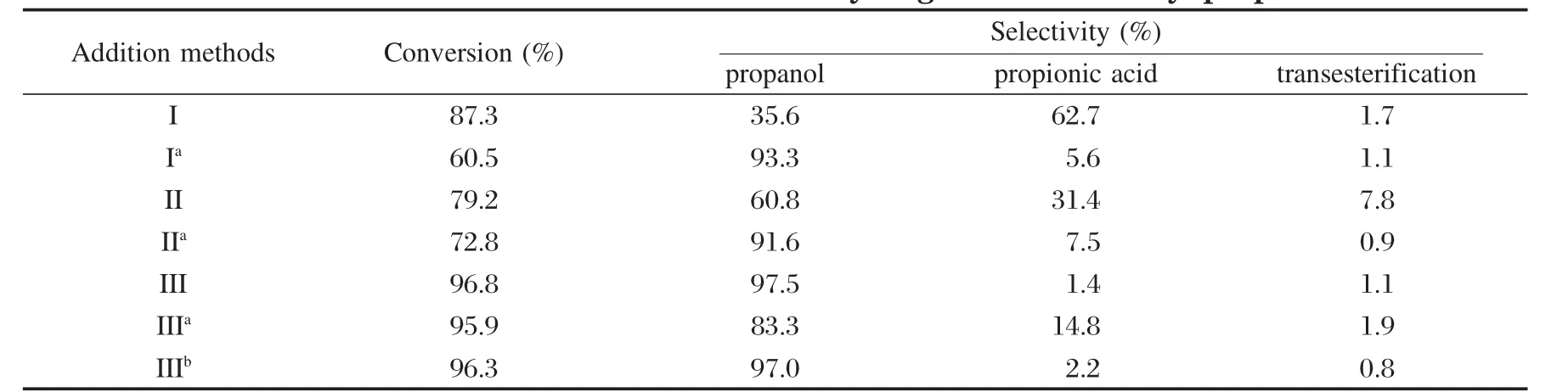
表3 Co不同添加方式对丙酸甲酯加氢的影响Table 3 Effect of Co2+addition method on hydrogenation of methyl propionate
表3数据显示,Co2+添加到溶液中的效果最好,而方法I和II都使催化活性降低.比较I和Ia、II和IIa发现,通过煅烧引起了催化剂活性明显降低,选择性也大幅下降.不经过煅烧,催化剂的活性下降,选择性下降不明显.由此可见,将Co2+全部吸附到催化剂上对酯加氢反应并没有促进作用.这些结果表明,经煅烧后Co(NO3)2将转化为氧化钴;将Co(NO3)2吸附在催化剂上不煅烧,钴将仍以离子态存在于催化剂表面.虽然氧化钴存在时,底物转化率比离子态的Co2+存在的催化剂高,但底物主要转化成了丙酸,生成目标产物的量很少.大量离子态的Co2+存在于催化剂表面时,催化剂的活性和选择性也没有改善.
由于Co2+被添加到水溶液中表现出最好的性能,为了研究清楚Co2+是在溶液中还是吸附在催化剂表面起作用,我们将三种方式反应后的溶液分别进行ICP分析,发现方式I和II反应后的滤液中只有极少量的Co2+,99.8%的Co2+都保持在催化剂表面;IIa反应后溶液中钴含量经ICP分析为2.0%,可见在催化剂表面吸附的Co2+对反应没有促进作用.而III反应后的溶液中存在17%的Co2+,有83%的Co2+吸附到了催化剂表面.我们又将III反应后的催化剂分离并再一次反应:一种是直接使用III反应后的催化剂(IIIa),不补加Co(NO3)2;另一种(IIIb)是用III反应后的催化剂,再补加17%的Co(NO3)2(与III反应后溶液中的Co2+量相同).从表3结果可以看出,在反应液中补加Co(NO3)2比不补加对生成丙醇的选择性要高.IIIa反应的溶液中有5.7%的Co2+存在,这是催化剂上吸附的Co2+再次进入溶液的缘故. IIIb反应后的滤液中存在10.2%的Co2+,比IIIa反应后溶液中的Co2+多了4.5%.从上述结果看出,溶液中Co2+量多,转化率和选择性则明显提高.上述结果说明了Co2+对反应的促进作用是发生在溶液中的.
另外,III反应后催化剂的XPS测试(图3)表明催化剂上没有金属态的Co,这说明在反应中是Co2+起促进作用.我们认为添加的Co2+也是作为路易斯酸.以水为溶剂时,丙酸甲酯在水中有一定的溶解性,溶于水中的Co2+和溶于水中的酯的羧基能发生配位作用,导致电负性大的氧原子上电荷密度下降,从而极化了C=O,促进了反应的进行.
2.4 电子因素、空间因素对酯加氢反应的影响
为了进一步证明羧基碳原子上正电荷密度增加有利于羧基的活化,我们以4%Ru-9%La/γ-Al2O3为催化剂,考察了底物电子因素、空间因素对反应的影响,结果列于表4.从表4中可以看出,随着底物分子中基团的增大(entry 1到entry 5),发现无论是羧酸链的增大,还是酯基的增大所引起的空间位阻都不利于底物在催化剂表面的吸附,因此加氢活性降低.再分别将entry 2和entry 6、entry 7和entry 8进行比较发现,将底物分子(entry 2和entry 7)中的—CH3变成—CF3(entry 6和entry 8)时,底物的转化率明显增加.由于—CF3基作为吸电子基团,—CF3的存在可以增加底物分子中羧基碳原子上的正电荷密度,同样起到了极化了C=O键的目的,因而有利于活化的氢进攻C=O键上的碳原子,使反应活性增强.

表4 催化剂对不同底物的加氢性能Table 4 Hydrogenation performance of the catalyst for different substrates
3 结 论
制备了Ru-La/γ-Al2O3催化剂,并用于丙酸甲酯的加氢反应,分别考察了溶剂、无机盐添加剂、底物的电子因素等对酯加氢的影响.发现以水为溶剂时,反应条件下γ-Al2O3转化成AlO(OH)、AlO(OH)的表面羟基与底物羧基氧形成的氢键可以引起羧基活化.而在反应溶液中添加适量的Co(NO3)2,钴作为路易斯酸能与酯的羧基氧发生配位作用,导致羧基氧原子上电荷密度下降,从而极化了C=O键.此外,底物分子中吸电子基团导致羧基碳原子上正电荷密度增加,同样有利于羧基的活化.因此羧基C=O键的极化或碳原子正电荷的增加是引起酯加氢活性改善的重要因素.
1 McAlees,A.J.J.Chem.Soc.C,1969:2425
2 Rieke,R.D.;Thakur,D.S.;Roberts,B.D.;White,G.T.J.Am.Oil Chem.Soc.,1997,74(4):333
3 Zhu,J.F.Preparation and characterization of middle-pressure hydrogenation catalyst for fatty acid methyl esters[D].Qingdao: China University of Petroleum(East China),2007 [朱建锋.脂肪酸甲酯中压加氢催化剂制备与表征[D].青岛:中国石油大学(华东),2007]
4 Luo,G.;Yan,S.R.;Qiao,M.H.;Zhang,J.H.;Fan,K.N.Appl. Catal.A:Gen.,2004,275:95
5 Bournonville,J.P.;Mabilon,G.;Candy,J.P.;Basset,J.M.J.Mol. Catal.,1991,67:283
6 Corradini,S.A.S.;Lenzi,G.G.;Lenzi,M.K.;Soares,C.M.F.; Santos,O.A.A.Journal of Non-Crystalline Solids,2008,354: 4865
7 Silva,A.M.;Morales,M.A.;Baggio-Saitovitch,E.M.;Jordão,E.; Fraga,M.A.Appl.Catal.A-Gen.,2009,353:101
8 Piccirilli,A.;Pouilloux,Y.;Pronier,S.;Barrault,J.Bull.Soc. Chim.France,1995,132:1109
9 Pouilloux,Y.;Autin,F.;Barrault,J.Catal.Today,2000,63:87
10 Silva,A.M.;Santos,O.A.A.;Morales,M.A.;Baggio-Saitovitch, E.M.;Jordão,E.;Fraga,M.A.J.Mol.Catal.A,2006,253:62
11 Santos,S.M.;Silva,A.M.;Jordão,E.;Fraga,M.A.Catal.Today, 2005,107-108:250
12 Miyake,T.;Makino,T.;Taniguchi,S.I.;Watanuki,H.;Niki,T.; Shimizu,S.;Kojima,Y.;Sano,M.Appl.Catal.A-Gen.,2009, 364:108
13 Pouilloux,Y.;Autin,F.;Guimon,C.;Barrault,J.J.Catal.,1998, 176:215
14 Zhou,Y.F.;Fu,H.Y.;Zheng,X.J.;Li,R.X.;Chen,H.;Li,X.J. Catal.Commun.,2009,11:137
15 Hu,S.C.;Chen,Y.W.J.Chem.Technol.Biotechnol.,2001,76: 954
16 Okada,K.;Nagashima,T.;Kameshima,Y.;Yasumori,A.; Tsukada,T.J.Colloid Interface Sci.,2002,253:308
17 Chen,X.Y.;Lee,S.W.Chem.Phys.Lett.,2007,438:279
18 Wang,J.Q.;Wang,Y.Z.;Xie,S.H.;Qiao,M.H.;Li,H.X.;Fan, K.N.Appl.Catal.A,2004,272:29
19 Fleisch,T.H.;Hicks,R.F.;Bell,A.T.J.Catal.,1984,87:398
20 Struijk,J.;Scholten,J.J.F.Appl.Catal.A,1992,82:277
21 Wehner,P.S.;Gustafson,B.L.J.Catal.,1992,135:420
22 Clapham,S.E.;Hadzovic,A.;Morris,R.H.Coord.Chem.Rev., 2004,248:2201
23 Nomura,K.;Ogura,H.;Imanishi,Y.J.Mol.Catal.A,2001,166: 345
24 Zuo,B.J.;Wang,Y.;Wang,Q.L.;Zhang,J.L.;Wu,N.Z.;Peng, L.D.J.Catal.,2004,222:493
25 Xu,Q.;Liu,X.M.;Chen,J.R.;Li,R.X.;Li,X.J.J.Mol.Catal.AChem.,2006,260:299
April 29,2010;Revised:June 3,2010;Published on Web:July 15,2010.
Activation Factors for the Carboxyl Group in the Hydrogenation of Carboxylic Esters
ZHENG Xiao-Juan ZHOU Ya-Fen FU Hai-Yan CHEN Hua LI Xian-Jun LI Rui-Xiang*
(Key Laboratory of Green Chemistry and Technology,Ministry of Education,College of Chemistry,Sichuan University, Chengdu 610064,P.R.China)
We prepared a 4%Ru-9%La/γ-Al2O3catalyst by impregnation method and characterized it using X-ray diffraction(XRD),X-ray photoelectron spectroscopy(XPS)and transmission electron microscopy(TEM).The catalyst was used for the hydrogenation of methyl propionate.The effects of solvent,inorganic salt additive,steric as well as electronic factors of the substrate on the hydrogenation of a carboxylic ester were investigated.We found that both water and the Co(NO3)2additive obviously improved the hydrogenation of methyl propionate,the conversion of the substrate and the selectivity for propanol.The promotional effects of water and Co2+are attributed to polarization of the C=O bond in the carboxyl group of the substrate molecule by the formation of a hydrogen bond between water and the carboxylic group and the coordination of Co2+to the carboxylic group.This is favorable for an attack on the carbon atom of the carboxyl group by the activated hydrogen.Similarly,the electron-withdrawing group in the substrate molecule also caused the high positive charge of carbon in the carboxyl group.The highly positive charged carbon is beneficial for the hydrogenation reaction.In addition,an increase in the steric hindrance of the substrate molecules was not favorable for the adsorption of the substrate on the catalyst and,therefore,the reaction rate decreased.
Catalytic hydrogenation;Methyl propionate;Activation;Ruthenium;Lanthanum
O643
*Corresponding author.Email:sculiruixiang@163.com;Tel:+86-28-85412904.
The project was supported by the National Natural Science Foundation of China(21072138).
国家自然科学基金(21072138)资助项目
猜你喜欢
云南化工(2020年11期)2021-01-14 00:50:52
现代食品(2016年24期)2016-04-28 08:12:06
化工进展(2015年3期)2015-11-11 09:07:41
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国当代医药(2015年10期)2015-03-01 02:02:39
海军医学杂志(2015年2期)2015-02-27 13:47:35
应用化工(2014年3期)2014-08-16 13:23:50
应用化工(2014年10期)2014-08-16 13:11:29
应用化工(2014年7期)2014-08-09 09:20:23
华东理工大学学报(自然科学版)(2014年6期)2014-02-27 13:49:40
