
A1腺苷受体的同源模建及其结构验证
2010-11-30 10:50:10柯艳蓉金宏威刘振明张亮仁
物理化学学报 2010年10期
柯艳蓉 金宏威 刘振明 张亮仁
(北京大学医学部药学院,天然药物及仿生药物国家重点实验室,北京 100191)
A1腺苷受体的同源模建及其结构验证
柯艳蓉 金宏威 刘振明 张亮仁*
(北京大学医学部药学院,天然药物及仿生药物国家重点实验室,北京 100191)
采用同源模建的方法构建了A1腺苷受体的三维结构,并与拮抗剂分子DPCPX对接,将得到的复合物结构进行5 ns的分子动力学模拟,以最后2 ns的平均结构和平衡后抽取的11帧构象共12个蛋白结构为研究对象,用包含52个活性分子和1000个诱饵分子的测试库,分别通过DOCK、VINA和GOLD三种对接软件进行评价,最终得出合理的蛋白质模型.根据top10%的富集因子(EF)和ROC曲线下面积(AU-ROC)的计算结果,我们认为GOLD是最适合A1腺苷受体的对接软件,而12个蛋白质结构中F5和Favg的三维结构模型比较合理,可以作为进一步大规模虚拟筛选的模型.
分子动力学模拟;A1腺苷受体; 同源模建;GOLD; 虚拟筛选
腺苷受体是G蛋白偶合受体(GPCR)蛋白受体家族成员,其主要结构特征包括了七次跨膜的疏水螺旋区(transmembrane,TM1-TM7)、N端区、三个胞外LOOP区(extracellular loop,EL1-EL3)、三个胞内LOOP区(intracellular loop,IL1-IL3)和C端区.胞内IL3和C端区与G蛋白偶联,从而影响环磷酸腺苷酶或磷酸脂酶C的活性,使细胞内产生第二信使,并通过信息传递,参与细胞物质代谢的调节和基因转录的调控.腺苷受体有四种亚型,分别为A1、A2A、A2B、A3[1],并且通过内源性的腺苷调节多种生理功能.腺苷受体有其独特的药理学特征、组织分布和偶联蛋白,作为药物靶标,其应用领域相当广泛,主要可应用于局部缺血性疾病(大脑和心脏)、睡眠功能障碍、免疫功能和炎症功能紊乱、癌症等的治疗.目前,已经设计合成出一系列的腺苷受体激动剂和拮抗剂,并且有部分化合物已进入临床研究.
A1受体主要作用于中枢神经系统和外周循环系统,并且对机体有促进免疫的作用.A1受体集中分布在脑和脊髓中,主要是在海马,小脑,上丘脑,皮质I、IV、VI中大量存在.另外,A1受体也广泛分布于心脏,在肾脏、肺和膀胱、脂肪等组织细胞中也有表达.A1受体作为潜在的药物靶标[2-3],吸引了很多的药物工作者对它的研究.因为,一方面A1受体的拮抗剂主要应用于抗高血压药、钾离子保留的利尿药、认知功能增强药、缓和阿尔兹海默综合症[4-5],也可用于治疗痴呆、缓解焦虑[6],以及治疗充血性心率衰竭患者的急性肾功能紊乱[7-8]等;另一方面,A1受体拮抗剂的发展可作为蛋白受体药理学特征的分子探针.该受体拮抗剂分子设计主要以天然的咖啡因、茶碱为母核结构进行化学修饰,主要有两种类型:黄嘌呤结构衍生物和非黄嘌呤结构衍生物(多聚杂环衍生物).目前,A1受体的晶体结构尚未解析出来,所以通常采用同源模建的方法构建该受体的模型.目前,已报道A1受体的模型,主要是以牛视紫红质受体为模板[9-11].2007年,β2-肾上腺素受体晶体结构被解析出来后[12],Yuzlenko等[13]分别以牛视紫红质受体和β2-肾上腺素受体为模板进行A1受体的同源模建,发现β2-肾上腺素受体更适合于腺苷受体的建模.2008年,Jaakola等[14]解析了A2A受体的晶体结构,给腺苷受体家族其他亚型受体的模建提供了更好的模板.
本文通过同源模建的方法模拟了A1受体的三维结构,将得到的模型与DPCPX拮抗剂进行分子对接,得到受体-配体复合物结构.将该复合物置于磷脂双分子层中进行分子动力学模拟,使受体模型结构得到优化.将包含52个A1受体拮抗剂的测试库分别与动力学平衡过程中抽取的11个构象和平均结构进行对接,以活性分子的top10%的富集因子(enrichment factor,EF)和ROC曲线下面积(AU-ROC)大小作为结果的评价依据,以便为进行大规模虚拟筛选提供合理的受体结构.
1 计算方法
A2A受体(分辨率0.26 nm,PDB编号3EML)结构数据来源于PDB蛋白晶体结构数据库[15].A1受体的氨基酸序列来源于SWISS-PROT数据库(登记号为P30542)[16].所有蛋白模建、模型优化、分子对接、动力学模拟计算均在DELL PowerEdge 2950(Xeon E5410-2.33G×2,内存8.0G)计算机工作站完成,所用程序为Sybyl 6.9(Tripos公司)和Discovery Studio (DS)2.1(Accelrys公司)分子设计软件包[17-18],计算中选用的各项参数除特别说明外均使用缺省值.
1.1 同源模建
A1受体由326个氨基酸残基组成.序列比对采用DS 2.1中的Align123程序,并根据A型G蛋白偶联受体家族中高度保守的氨基酸残基位置对结果进行手动调整.采用DS 2.1中的MODELER程序对A1受体模型进行搭建,使用BLOSUM矩阵,多序列断点罚分(gap open penalty)为5.0,多序列断点伸展罚分(gap extension penalty)为0.05.由程序自动生成10个模型,选取概率密度函数(probability density functions,PDF)对蛋白质几何性质打分最高的模型,用Procheck程序进行合理性评价.
1.2 分子对接
首先使用SYBYL 6.9软件对小分子和蛋白进行处理.A1受体拮抗剂DPCPX分子的初始结构使用sketch模块构建,如图1所示,并对小分子加氢和加Gasteige-Hückel电荷.对所构建的初始结构依次进行1000步最陡下降法和1000步共轭梯度法优化.使用Biopolymer模块对蛋白模型加极性氢和赋予AMBER7 FF99电荷.分子对接使用GOLD 3.0.1程序[19],将DPCPX分子置入受体的活性位点.我们根据文献报道的突变数据[20-21]来定义A1受体的活性位点,即以Ser94和His278两个关键残基的中心坐标为中心,半径为2.5 nm的球形空间为活性口袋.该口袋包含的关键残基有Leu88、Thr91、Gln92、Ser94、His251和His278[22].采用遗传算法(GA)进行蛋白-配体对接,对接时考虑配体柔性和蛋白部分柔性.采用Goldscore打分函数,选择打分值最高的小分子结合构象进行动力学研究.

图1 DPCPX化学结构Fig.1 Chemical structure of DPCPX
1.3 分子动力学模拟
对于DPCPX分子使用Gaussian 03程序[23],用HF方法,在6-31G*基组水平下计算该分子的静电势.然后用AMBER 8.0软件包中的Antechamber程序,用RESP方法计算DPCPX的部分原子电荷[24],得到小分子力场文件.MD模拟使用Gromacs 3.2.1程序包[25-26],用‘genbox’命令将对接得到的复合物置入DPPC256膜中.膜中心56个磷脂分子被自动删除避免与蛋白产生碰撞.然后,对整个体系添加SPC水模型,添加13个氯离子,使体系保持电中性.模拟体系共有58147个原子,其中3078个蛋白质原子,200个磷脂膜分子,15011个水分子,13个氯离子.
采用Gromacs 96[27]力场,将蛋白质复合物-膜-水体系分别用最陡下降法和共轭梯度各进行1000步的能量优化,以避免模拟时原子间的不合理碰撞;然后,束缚蛋白和小分子,对膜-水体系进行200 ps的动力学模拟;再束缚蛋白主链和小分子,对蛋白侧链-膜-水体系进行200 ps的动力学模拟;最后对整个体系进行5 ns的恒温恒压分子动力学模拟计算.动力学过程中,步长设为2 fs;使用Berendsen等[28]提出的温度和压力耦合方法,使系统温度保持在310 K,耦合时间0.1 ps;压力保持在105 Pa,采用0.5 ps耦合时间模拟自由水.模拟水分子的等温压缩系数设为 4.5×10-10Pa-1,用 LINCS(linear constraint)算法[29]约束所有原子的键长,计算距离小于1 nm的带电基团之间的静电相互作用;用PME(particle-mesh Ewald)方法[30]计算长程静电相互作用,格点宽度设为0.12 nm;Lennard-Jones相互作用的截断距离设为1 nm.
在最后2 ns模拟时间内,每隔20帧保留一个构象,总共抽取了11个构象,分别命名为F1-F11.再计算得到最后2 ns模拟时间内的平均结构,并对该结构进行真空中的800步最陡下降法和1200步的共轭梯度法的能量优化,获得最终结构命名为Favg.这12个DPCPX-A1受体的复合物结构将用于进一步模型评价.
1.4 小分子数据库的制作
从MDDR(Elevier MDL,San Leandro CA)数据库中,随机抽取52个A1受体的拮抗剂,与来自薛定谔网站上的1000个分子量为400的decoys,组成一个含有1052个分子的测试库.使用Concord v 6.1.3程序得到测试库分子的三维结构用于虚拟筛选评价.
1.5 模型评价
虚拟筛选评价采用VINA、DOCK 4.0、GOLD 3.0.1三种对接软件.使用DOCK 4.0软件包[31-32]进行对接时,通过Sphgen程序对蛋白负像进行聚类,以每个复合物结构中的配体分子定义活性位点,格点球半径为1 nm.对接结果以能量打分进行排序.使用VINA软件[33]进行对接,受体和小分子先后在MGLTools 1.4.6中打开进行对接前文件处理,对于受体结构,添加极性氢原子,计算电荷.用小分子定义活性位点的中心,并界定2 nm×2 nm×2 nm的对接格点范围.对接结果进行能量排序.使用GOLD 3.0.1软件[19]进行对接,以配体小分子定义活性位点,以小分子为中心半径为2.5 nm的球形空间为活性口袋.采用遗传算法(GA)进行蛋白-配体对接,对接时考虑配体柔性和蛋白部分柔性.采用Goldscore打分作为打分函数,以GOLD fitness function值进行排序.
2 结果与讨论
2.1 同源模型的评价
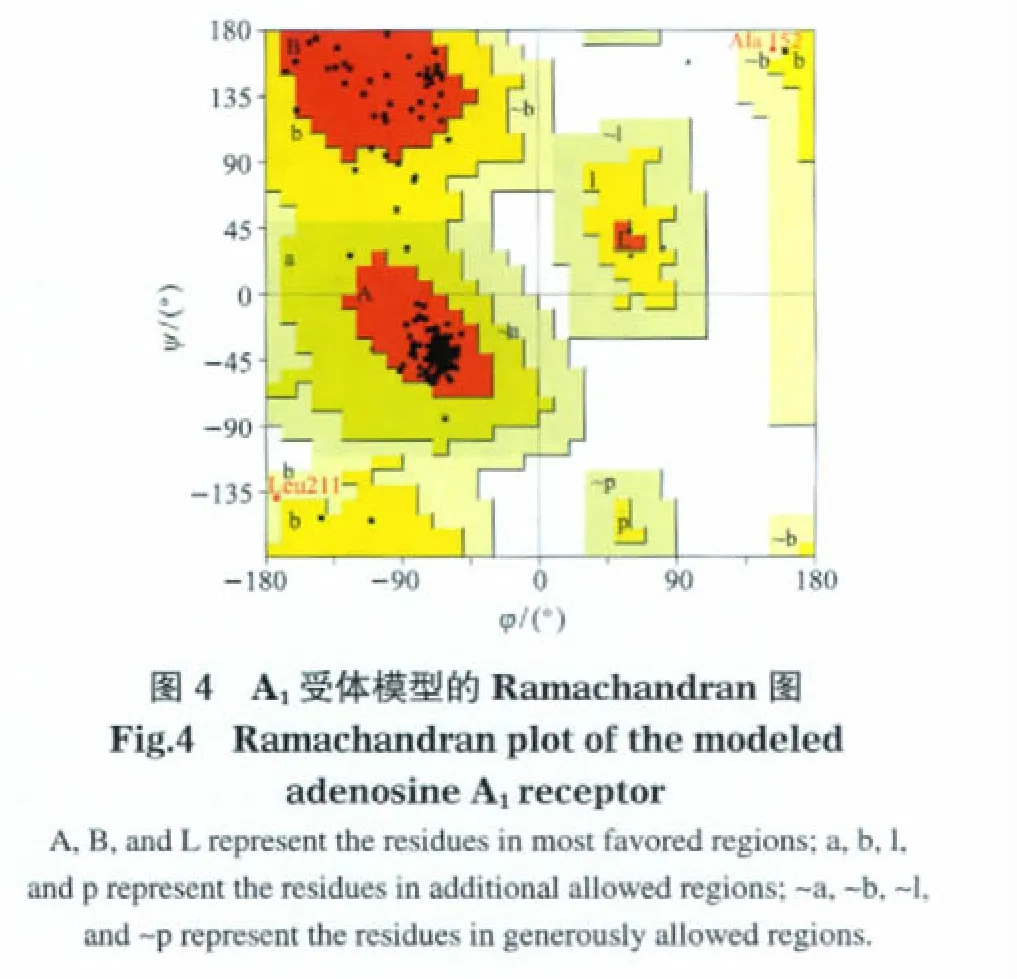
通过Align123程序的序列比对如图2所示,结果表明A1和A2A具有较高的序列相似度(similarity:71.6%)和一致性(identity:54.1%),是以其他GPCRs蛋白晶体结构为模板的多序列比对达不到的.而且,模建模型中保留了腺苷受体TM3和EL2二硫键(A1:Cys80-Cys169)的特征,且EL3中Cys260-Cys263二硫键特征(见图3)与A2A晶体结构二硫键特征相符[14].我们使用Procheck程序对模型结构的立体化学参数进行了评价,该程序可以凭经验比较给定蛋白质结构与最合理的蛋白质结构之间立体化学性质的差异.评价结果中主要考察所有氨基酸残基骨架的二面角分布,并通过程序生成的Ramachandran图来表示(见图4).我们可以看到模建的蛋白质有93.5%的残基落入最佳区域(the most favoured regions),5.8%的残基落入其它许可区(additional allowed regions),0.7%的残基落入勉强许可区(gener-ously allowed regions),无残基落入不允许区(disallowed regions).上述结果说明模建得到的A1受体模型氨基酸二面角结构是比较合理的.


2.2 受体抑制状态模型的构建和分子动力学模拟
DPCPX是黄嘌呤结构的高选择A1受体的拮抗剂分子,该分子与A1受体的亲和力 Ki值为3.9 nmol·L-1,其选择性表现为该分子与其它亚型受体的亲和力 Ki值分别为 A2A:129 nmol·L-1、A2B:56 nmol·L-1、A3:3980 nmol·L-1[34].而且,同位素[3H] DPCPX被广泛应用于A1和A2B受体放射性结合实验和突变实验研究.因此,我们选择DPCPX分子与A1模型进行分子对接.选择Goldscore打分最好的构象,进行下一步的动力学模拟.
将A1受体-DPCPX复合物模型置于DPPC膜中进行了5 ns的分子动力学模拟(图5).在模拟过程中,DPCPX小分子拮抗剂诱导大分子受体构象逐渐变化,最终达到能量平衡.相对于起始结构的均方根偏差(RMSD)计算结果证明了轨迹的稳定性.图6给出了5 ns模拟时间内蛋白质骨架和小分子的RMSD值随时间的变化曲线.可以看到,在开始的1.5 ns模拟时间内,蛋白质骨架的RMSD值(黑色曲线)缓慢上升达到0.25 nm,表明了蛋白分子的运动和结构的不断变化.而在1.5 ns之后,RMSD值趋于稳定,在 0.25-0.3 nm之间上下波动.小分子的RMSD值(红色曲线)变化不大,一直稳定在0.1 nm范围内,处于平衡的动力学状态.从该体系在模拟过程中的势能变化曲线(图7)可以看出,体系的势能在最初的十几ps时间内迅速降低,然后随着模拟的进程势能逐渐降低,并从3 ns开始势能趋于稳定直到5 ns,说明该体系已经达到平衡.根据分子动力学模拟结果,我们从3 ns起每隔20帧取一个构象,共取得11个构象(F1-F11).再加上3-5 ns时间内的平均结构(Favg),共得到12个结构.
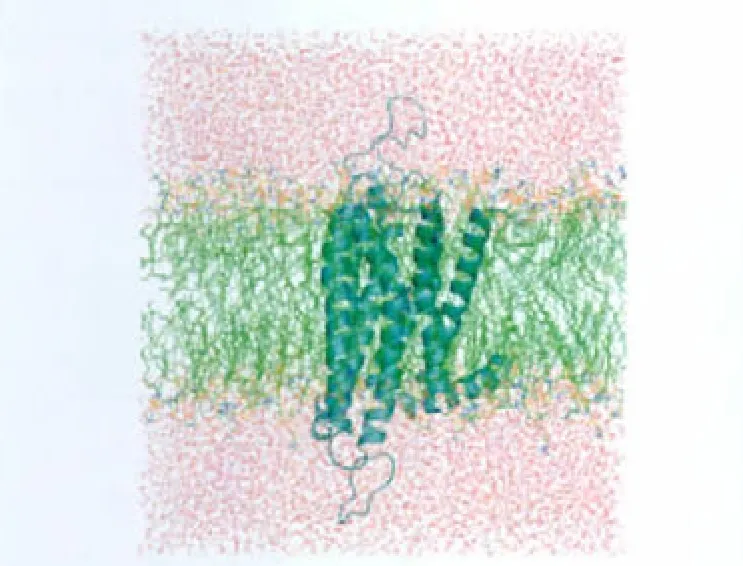
图5 动力学模拟过程中DPPC256磷脂膜-A1受体-DPCPX体系的初始结构模型Fig.5 Initial model containing DPPC256,A1 receptor,and DPCPX for the molecular dynamics simulation
图8给出了Favg中,小分子和蛋白的结合模式.我们可以看到,DPCPX分子主要结合区域是TM2、TM3、TM6、TM7,残基 His278、Val58、Val87和残基 Leu90、Thr91、Ala54、Asp55分别构成了两个疏水性口袋,与DPCPX分子的1位和3位的丙基有疏水作用;Leu51、Leu98、Asn284和Leu239残基也构成了疏水性口袋,与8位的环戊烷有疏水作用.此外,DPCPX的4位羰基与Asn280和Ser281肽键上的氨基形成氢键(氢键键长:0.2 nm),7位氨基氮与Ser94的羟基氧具有氢键相互作用(氢键键长:0.3 nm).根据文献报道,对于A1腺苷受体,突变Ser94和His278这两个氨基酸残基,则蛋白完全丧失与拮抗剂的结合.我们的对接结果与突变实验结果基本符合[20-22].

图6 5 ns动力学模拟过程中蛋白质骨架(a)和小分子拮抗剂DPCPX(b)的均方根偏差(RMSD)与时间的关系曲线Fig.6 Root mean square deviation(RMSD)vs time of the atoms from their initial positions during the 5 ns simulation of protein backbone(a)and DPCPX(b)
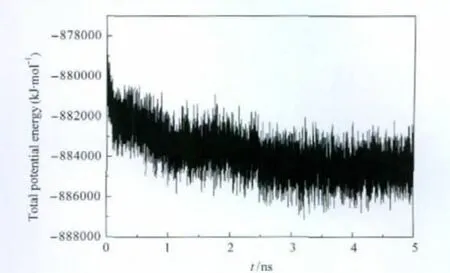
图7 动力学模拟过程中体系的势能变化Fig.7 Total potential energy changes of the system during molecular dynamics simulation
2.3 用测试数据库验证模型
采用DOCK 4.0、VINA和GOLD 3.0.1对接软件,将分子动力学模拟得到的12个蛋白质模型与测试库分子进行对接.用EF和AU-ROC两个参数来评价蛋白模型是否合理.
EF是评价虚拟筛选好坏的一个重要指标,它是指,在选择的排名前百分之几(例如10%)的数据集中活性分子的浓度与初始活性分子浓度的比值.这个指标用于评价筛选中筛选出活性分子的阳性率.富集因子计算公式为[35]:
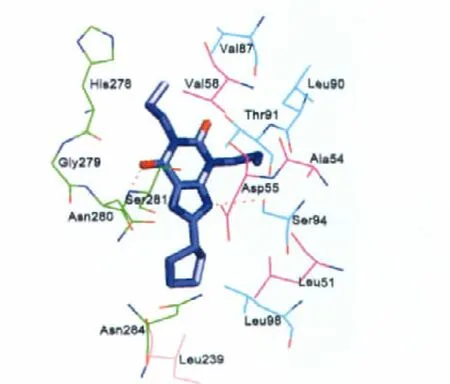
图8 DPCPX与A1受体结合模式Fig.8 Binding profile of DPCPX and adenosine A1receptorred:the residues in TM2;blue:the residues in TM3; orange:the residues in TM5;green:the residues in TM6

其中,ligandsselected指选择的排名前10%的分子中活性小分子的数量,Nsubset指选择的前10%的小分子的数量,ligandstotal指整个测试库中活性分子的总数,为52,Ntotal指整个测试库分子的总数,为1052.
ROC(receiver operator characteristic)曲线描绘的是敏感度(sensitivity,Se)和特异性(specificity,Sp)的关系.AU-ROC值指的是ROC曲线下的面积[36-37].这个指标用于测试虚拟筛选策略的优劣,当被评价的筛选策略都筛选出相同比例的诱饵分子时,筛出活性分子的比例越高说明该策略对活性分子的敏感度越高.AU-ROC值是以具体数值表示ROC曲线下的面积,该值的区间是[0,1],1表示筛出0个诱饵分子时筛出所有的活性分子;0表示筛出全部的诱饵分子中不含有活性分子.
因此,我们希望真正有活性的化合物能够尽可能富集在前百分之十比例(top10%)的筛选结果中,我们也希望AUC曲线下面积能越大越好,这样有利于评价这12个蛋白结构的优劣性,选择更合理的蛋白结构和更合适的对接软件用于进一步大规模虚拟筛选研究和小分子结合模式的研究.将12个蛋白质结构分别通过DOCK 4.0、VINA、GOLD 3.0.1三个对接软件分别与测试数据库对接后,对接结果用相应的评分函数打分并进行排序和计算,得到相关的测试库中52个活性分子的EF和AU-ROC值(表1).从表 1中 EF10和AU-ROC的均值,可看出DOCK、GOLD、VINA三种对接软件中,最适合A1受体进行虚拟筛选的对接软件是GOLD.而且,三种对接软件都可以特异性地选择某些构象的蛋白结构.对于DOCK软件更趋向于选择F8、F3、F4蛋白结构,VINA软件更趋向于选择F2、Favg、F6蛋白结构,而GOLD软件更趋向于选择F5、Favg、F4蛋白结构.
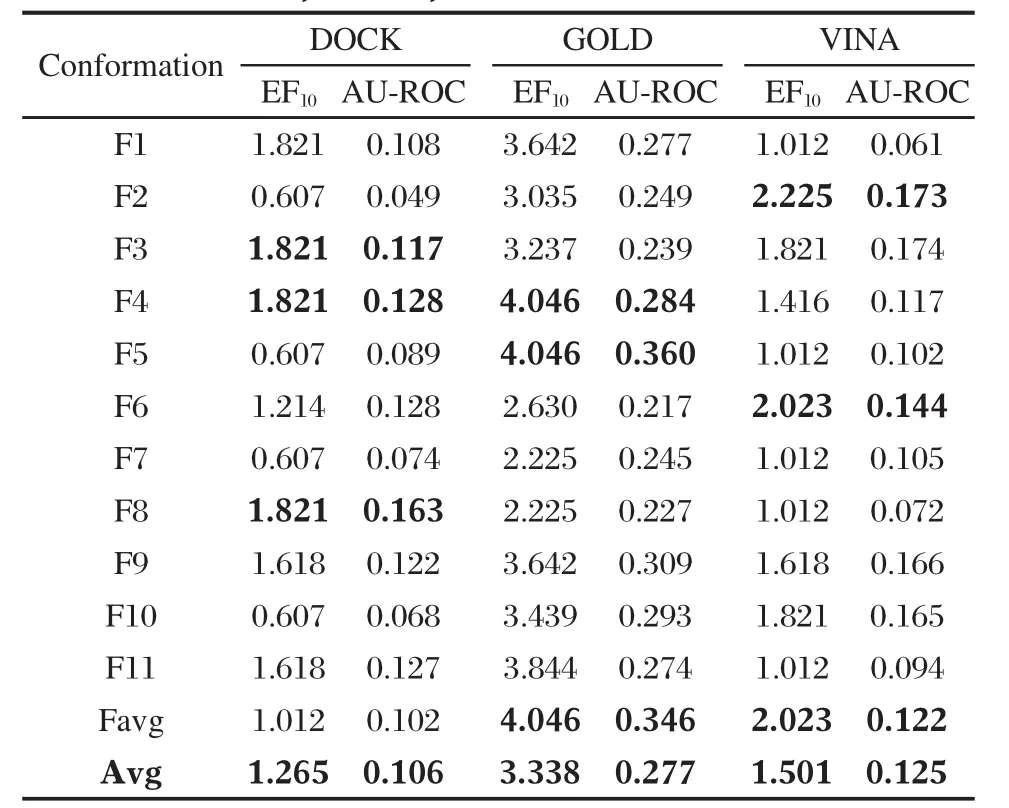
表1 DOCK、GOLD和VINA软件的对接评价结果Table 1 Evaluations of docking results obtained by DOCK,GOLD,and VINA softwares

表2 DOCK、GOLD和VINA软件配对t检验的结果Table 2 Results of DOCK,GOLD,and VINA softwares′paired samples test
分别以三个软件中各自评价最高和评价最低的两组蛋白模型对接计算中52个活性分子的对接能量值作为样本数据,利用SPSS软件进行了配对t检验统计学分析,在5%显著性水平的条件下,自由度为50的t临界值为3.261.选择的蛋白模型包括Favg和F8(GOLD)、F2和F1(VINA)以及F8和F2 (DOCK),分析结果见表2.可以看到,对于GOLD(t= 3.775,p<0.001)、VINA(t=5.717,p<0.001)和DOCK (t=8.495,p<0.001)计算所得的t值明显大于临界值,说明对接中各自的两组蛋白的小分子对接能量存在着显著性差异,即Favg蛋白的虚拟筛选能力比F8蛋白强(GOLD);F1蛋白的虚拟筛选能力比F2蛋白强(VINA);F8蛋白的虚拟筛选能力比F2蛋白强(DOCK).总之,DOCK、GOLD和VINA对接软件可以特异性地选择某些构象的蛋白质结构,并可以区分出蛋白模型的优劣.
应用GOLD软件进行虚拟筛选时,F4、F5和Favg的EF10值均为4.046,说明它们是12个蛋白质模型中比较合理的结构.此外,这三个蛋白质结构的曲线下面积(AU-ROC)分别为0.284、0.360、0.346,说明F5和Favg是两个不错的模型,可用于进一步的大规模虚拟筛选和小分子拮抗剂结合模式的研究.
3 结 论
用同源模建的方法对A1受体的三维结构进行构建,经过磷脂双分子膜中的分子动力学模拟,抽取了动力学平衡中最后2 ns的11个构象(F1-F11)和计算动力学平衡中的平均构象(Favg).用包含52个活性分子的测试库分别用DOCK、VINA、GOLD三种软件与12个模型进行对接以验证模型结构,从EF10和AU-ROC平均值结果分析表明GOLD软件是这三个软件中最适合A1受体虚拟筛选.而且,这三种软件都具有筛选出最优构象模型的能力,最优构象与最劣构象的小分子对接能量存在着显著性差异.从GOLD软件对12个蛋白模型的选择中,我们可知F5和Favg是最合理的蛋白结构模型,可进一步用于大规模虚拟筛选和小分子拮抗剂结合模式的研究.
1 Ralevic,V.;Burnstock,G.Pharmacol.Rev.,1998,50:413
2 Müller,C.E.;Stein,B.Curr.Pharm.Design,1996,2:501
3 Poulsen,S.A.;Quinn,R.J.Bioorg.Med.Chem.,1998,6:619
4 Müller,C.E.Expert.Opin.Ther.Pat.,1997,7:419
5 Hess,S.Expert.Opin.Ther.Pat.,2001,11:1533
6 Maemoto,T.J.Pharmacol.Sci.,2004,96:42
7 Wilcox,C.S.;Welch,W.J.;Schreiner,G.F.;Belardinelli,L. J.Am.Soc.Nephrol.,1999,10:714
8 Gottlieb,S.S.Circulation,2002,105:1348
9 Giordanetto,F.;Fossa,P.;Menozzi,G.;Schenone,S.;Bondavalli, F.;Ranise,A.;Mosti,L.J.Comput.Aided Mol.Des.,2003,17:39
10 Gutiérrez-de-terán,H.;Centeno,N.B.;Pastor,M.;Sanz,F. Proteins,2004,54:705
11 Tuccinardi,T.;Ortore,G.;Manera,C.;Saccomanni,G.;Martinelli, M.Euro.J.Med.Chem.,2006,41:321
12 Cherezov,V.Science,2007,318:1258
13 Yuzlenko,O.;Kiec-Kononowicz,K.J.Comput.Chem.,2009,30: 14
14 Jaakola,V.P.Science,2008,322:1211
15 http://www.gpcr.org
16 http://expasy.org/sprot/
17 SYBYL software.Version 6.9.St.Louis:Tripos.Associates.Inc.
18 Discovery Studio software.Version 2.1.San Diego:Accelrys.Inc.
19 Jones,G.;Willett,P.;Glen,R.C.;Leach,A.R.;Taylor,R.J.Mol. Biol.,1997,267:727
20 Barbhaiya,H.;McClain,R.;Ijzerman,A.;Rivkees,S.Mol. Pharmacol.,1996,50:1635
21 Olah,M.E.;Ren,H.;Ostrowski,J.;Jacobson,K.A.;Stiles,G.L. J.Biol.Chem.,1992,267:10764
22 Martinelli,A.;Tuccinardi,T.Mol.Res.Rev.,2008,28:247
23 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision B.03.Wallingford,CT:Gaussian Inc.,2003
24 Bayly,C.I.;Cieplak,P.;Cornell,W.D.;Kollman,P.A.J.Phys. Chem.,1993,97:10269
25 Berendsen,H.J.C.;van der Spoel,D.;van Drunen,R.Comp.Phys. Commun.,1995,91:43
26 Lindahl,E.;Hess,B.;van der Spoel,D.J.Mol.Model.,2001,7: 306
27 van der Spoel,D.;Buuren,A.R.V.;Tieleman,D.P.;Berendsen, H.J.J.Biomol.NMR,1996,8:229
28 Berendsen,H.J.C.;Postma,J.P.M.;van Gunsteren,W.F.; Dinola,A.;Haak,J.R.J.Chem.Phys.,1984,81:3684
29 Hess,B.;Bekker,H.;Berendsen,H.J.C.;Fraaije,J.G.E.M. J.Comput.Chem.,1997,18:463
30 Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.,1993,98: 10089
31 Kuntz,I.D.;Blaney,J.M.;Oatley,S.J.;Langridge,R.;Ferrin,T. E.J.Mol.Biol.,1982,161:269
32 Ewing,T.J.A.;Kuntz,I.D.J.Comput.Chem.,1997,18:1175
33 Trott,O.;Arthur,J.O.J.Comput.Chem.,2010,31:455
34 Moro,S.;Gao,Z.G.;Jacobson,K.A.;Spalluto,G.Med.Res.Rev., 2006,26:131
35 Wei,B.Q.;Baase,W.A.;Weaver,L.H.;Mattews,B.W.; Shoichet,B.K.J.Mol.Biol.,2002,322:339
36 Hevener,K.E.;Zhao,W.;Ball,D.M.;Babaoglu,K.;Qi,J.J.; White,S.W.;Lee,R.E.J.Chem.Inf.Model.,2009,49:444
37 Truchon,J.F.;Bayly,C.I.J.Chem.Inf.Model.,2007,47:488
May 19,2010;Revised:June 7,2010;Published on Web:July 14,2010.
Homology Modeling and Structure Validation of the Adenosine A1Receptor
KE Yan-Rong JIN Hong-Wei LIU Zhen-Ming ZHANG Liang-Ren*
(State Key Laboratory of Natural and Biomimetic Drugs,School of Pharmaceutical Sciences,Health Science Center, Peking University,Beijing 100191,P.R.China)
A three dimensional structure model of the adenosine A1receptor was built using homology modeling. The antagonist DPCPX was docked into the model protein to form a receptor-ligand complex.A molecular dynamics simulation over 5 ns was performed for this complex.We selected 12 protein structures,including the average structure obtained from the last 2 ns of the simulation and 11 frames extracted after equilibration for the study.A database comprising 52 active antagonists and 1000 decoys was then docked into the 12 protein models using DOCK,VINA, and GOLD software packages and these molecules were ranked by their docking scores.The best model protein with the highest enrichment factor(EF)and the largest area under the ROC(AU-ROC)was chosen for further study.The results from the enrichment factor at 10%of the ranked database(EF10)and AU-ROC calculations indicate that GOLD is the best virtual screening software for the adenosine A1receptor.GOLD docking results suggest that optimized adenosine A1receptor protein structures,Favg and F5,can be used for virtual screening and for novel design to discover more potent antagonists.
Molecular dynamics simulation;Adenosine A1receptor;Homology modeling;GOLD; Virtual screening
O641
*Corresponding author.Email:liangren@bjmu.edu.cn;Tel:+86-10-82802567.
The project was supported by the National S&T Major Project Foundation,China(2009ZX09501-002).
国家科技重大专项关键技术基金(2009ZX09501-002)资助项目
猜你喜欢
空气动力学学报(2022年4期)2022-08-23 06:51:26
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
海南医学(2016年8期)2016-06-08 05:43:00
中国社区医师(2015年14期)2015-12-24 00:37:31
浙江大学学报(工学版)(2015年2期)2015-05-30 07:04:53
生殖医学杂志(2015年11期)2015-02-28 16:32:16
中华皮肤科杂志(2014年4期)2014-12-19 12:55:43
应用化工(2014年7期)2014-08-09 09:20:23
火炸药学报(2014年1期)2014-03-20 13:17:22
计算物理(2014年2期)2014-03-11 17:01:51
