
一种一枝黄花内酯分子结构与构象的计算研究
2016-10-27 14:57朱天文
温州大学学报(自然科学版) 2016年1期
朱天文
(云南大学化学科学与工程学院,云南昆明 650500)
一种一枝黄花内酯分子结构与构象的计算研究
朱天文
(云南大学化学科学与工程学院,云南昆明 650500)
一枝黄花内酯是一种从一枝黄花植物中提取到的克罗烷二萜类天然产物,对许多昆虫具有拒食活性.构建了Solidagolactone (4)分子的4种不同的空间结构,利用密度泛函理论(DFT)计算各个构象的能量值,找到相对稳定的两种构象进行化学位移值计算.研究表明,支链的构象会影响分子的稳定性;通过计算得到的化学位移值与实验值存在的误差,可引入校正因子对结果进行修正.
一枝黄花内酯;克罗烷二萜;分子构象;化学位移;支链
一枝黄花(Solidago decurrens Lour.)是一种菊科植物,可用于治疗慢性肾炎、膀胱炎、结石、风湿、糖尿病等疾病,具有抗氧化、抗菌等药理作用[1].其化学成分比较复杂,主要是二萜、黄酮类化合物.一枝黄花内酯是从该植物中提取到的一种天然产物,属于克罗烷二萜(Clerodane Diterpenes)范畴.
一般将克罗烷二萜分为两大类[2]:克罗烷型(Clerodane)(图1中A和B所示)和对映-克罗烷型(ent-Clerodane)(图1中C和D所示).这两大类中均包含顺式(trans-)和反式(cis-)两种构象,其主要区别在于C5与C10的取代基朝向是否在同一侧.此外,这两大类克罗烷二萜在相同顺反式的条件下互为对映异构体,即C5、C8、C9、C10的立体化学刚好相反.

图1 克罗烷二萜的基本构象
本文研究选择了一种一枝黄花内酯分子,其平面结构如图2[3]所示.
显然,该分子属于对映-反式-克罗烷类化合物,拥有较长的支链以及一个不饱和的内酯环.Enriz等[4]通过克罗烷二萜对黄粉虫的拒食活性研究指出,该类化合物侧链上的呋喃环和α,β-不饱和羰基(或环氧螺环)结构片段是拒食活性必不可少的.Solidagolactone (4)分子的支链上恰好含有类似的结构,因而可以推测出该分子有一定的拒食活性.此外,支链空间取向的不同会影响分子的稳定性,从而可能进一步影响分子的生物活性,所以对Solidagolactone (4)分子结构的解析显得很有必要.
构建出Solidagolactone (4)分子的4种拥有不同支链构型的三维结构,通过密度泛函理论(DFT)并使用Gaussian 09软件对于这4种构象进行优化,得到各个构象的能量.通过不同构象间的平行比较可确定出几种相对稳定的构象,并预测出这些构象是该分子中的主要构象.对于这几种构象进行13C NMR化学位移值的计算,并与实验值进行比较,通过计算值与实验值的拟合直线引入校正因子,即可确定出理论计算的方法对于该分子解析的可行性.

图2 Solidagolactone (4)平面结构与碳原子编号
1 计算方法
实验所有的DFT计算均通过Gaussian 09软件包实现.首先,利用GaussView 5.0.9构建目标分子的4种不同空间构象,再调用Becke型3参数密度泛函模型、Lee-Yang-Parr泛函(B3LYP),6-31G(d,p)基组在以氯仿为溶剂的环境中对各个构象进行结构优化与振动频率计算,最终得到势能最低的4种构象.对于13C NMR化学位移值的计算,使用Gauge-Independent Atomic Orbital(GIAO)方法[5],参照文献中的天然产物的化学位移计算方法,调用MPW1PW 91密度泛函模型[5],6-311+G(2d,p)基组[5],同样选择氯仿构造溶剂环境,对已优化的构象进行计算.此外采用四甲基硅烷(TMS)为基准物,设置上述条件进行结构优化得到化学位移基准值,为187.316 7 ppm.

图3 Solidagolactone (4) 分子的4种构象
2 结果与讨论
2.1 分子结构与构象的性质
如图3所示为Solidagolactone (4)分子的四种无虚频的稳定空间构象.显然,这四种构象可通过分子内单键的旋转相互转化得到.构象1为分子的平面结构通过Chem 3D软件构建,在4种构象中可认为是最原始的构象.旋转构象1中的C12-C13单键,可得到构象2.再旋转构象2中的C11-C12单键,可得到构象3.同理,旋转构象3的C12-C13单键得到构象4,而构象4则也可以通过构象1直接旋转C11-C12得到.4种构象的支链结构有明显不同,导致了能量与部分键角角度存在差异.Solidagolactone (4)分子的4种构象的支链结构见图4.

图4 Solidagolactone (4)分子的4种构象的支链结构
通过图4发现,构象1与构象4拥有相同的内酯环结构,而构象3和构象4拥有相同的除内酯环外的链状结构,这也就导致了4种构象在某些键角角度上的两两相似的情况.表1反映了4种构象通过计算得到的各项参数的数值情况.在结构的能量方面,若以构象1的能量为零点,可得出构象2、构象3和构象4的能量相对值.通过比较发现,构象1和构象2能量非常接近,这是由于内酯环空间结构的微小差异导致了能量的不同,但相较于构象3和构象4而言要稳定很多,因此可以预测出Solidagolactone (4)分子主要是以构象1和构象2存在的.而构象3与构象4的内酯环由于非常靠近结构的母环,有很强的空间位阻效应,结构的能量迅速升高.构象4的内酯环具有与构象1相同的空间取向,因而能量值略低于构象3.在偶极矩方面,4种构象的偶极矩非常接近.在键角角度方面,出现了两两相似的情况.例如,构象3与构象4都有C11-C12的旋转,所以两种构象各自的∠C11-C12-C13数值较为接近,∠C9-C11-C12亦是如此.同理,构象2与构象 3的内酯环有相同的空间取向,因此两种构象各自的∠C12-C13-C14数值也非常接近,∠C12-C13-C16亦是如此.

表1 Solidagolactone (4)分子的4种构象的能量、偶极矩和键角
2.213C NMR化学位移值
为了进一步对结构展开解析工作,利用Gaussian 09软件包对于构象1和构象2进行13C NMR化学位移值计算,并结合Müller等[3]利用CDCl3为溶剂,TMS作为基准物的实验化学位移值,得到表2.对照实验化学位移值,注意到,C15受到羰基的强吸电子作用,屏蔽效应减弱,化学位移值应该为最大,而实验值符合这一点,然而在计算中2种构象的计算值均显示C13的化学位移值最大,显然计算与实验在分析结果上存在很大不同.

表2 构象1和构象2的13C NMR的计算值与实验值
可将计算值与实验值的差值以柱形图的形式反映出来,如图5所示.
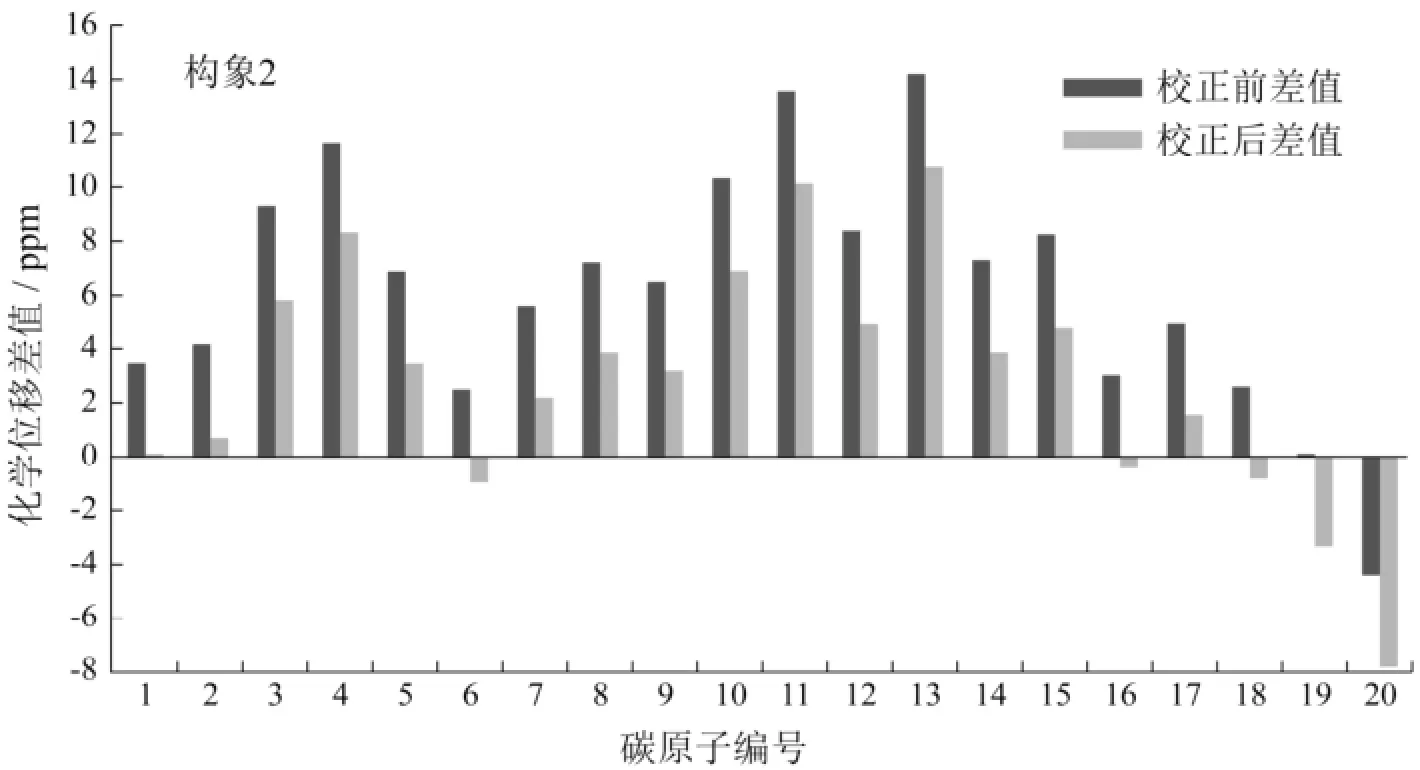
图5 构象1和构象2的13C NMR化学位移计算值与实验值之差
通常计算值与实验值的差值往往只有几个ppm,而图5中所示构象1和构象2均有碳原子的化学位移差值超过12 ppm的情况,比如构象1的C13,构象2的C11和C13.目前的结论并不让人满意.此时可人为引入一个校正因子来提高结果的精确度,即将表2中每种构象计算值为纵坐标,实验值为横坐标进行线性拟合.在极限情况下,若实验值为0 ppm时,计算值也应为0 ppm,因而拟合直线的截距可作为一个校正因子引入,同时直线的斜率也能为计算机模拟核磁共振谱的可行性提供依据.图6为构象1和构象2的13C NMR化学位移计算值与实验值拟合直线.

图6 构象1和构象2的13C NMR化学位移计算值与实验值拟合直线
图6反映了以构象1和构象2各自的13C NMR的计算值为纵坐标,实验值为横坐标,利用Origin Pro 9.0软件得到的一组拟合直线.以构象1为例分析,如图所示,该拟合直线的方程为y=3.004 62+1.048 4x(R2=0.996 32),线性良好.因此,该直线方程的截距3.00 462 ppm可作为一个校正因子引入,从而得到了校正后的计算与实验化学位移差值(Δδ值),如图5中构象1的灰色系列所示.不难发现,原本有部分差值在12 ppm左右,经过校正后,除了存在明显系统误差的C13,整体的差值在8 ppm左右,效果明显.此外,该拟合直线的斜率为1.048 4≈1,证实了通过计算方法得到的化学位移值的合理性,但是显然核磁计算的泛函模型对于该分子并不是最合适的,有待于进一步的研究.
同理,构象2的拟合方程为y=3.406 15+1.047 68x(R2=0.995 4),最终经过校正,同样除去存在明显系统误差的C13,整体的差值在9 ppm左右.相比之下,校正后的化学位移差值构象1比构象2整体更小一点,可以预测出Müller等[3]通过全合成得到的Solidagolactone (4)分子中主要有构象1和构象2,构象1所占比例略大.
3 结 论
通过对一种一枝黄花内酯分子—— Solidagolactone (4)结构的解析,找到了该分子的几种稳定构象,讨论了不同支链构象对分子的影响以及利用计算化学软件得到了13C NMR的化学位移计算值.显然,计算软件的系统误差无法避免,此外模拟的氯仿溶剂环境与实验条件下氘代氯仿的溶剂环境也存在一定的差异,通过引入校正因子对计算值进行合理修正能够得到令人满意的结果.计算化学方法的优势在于能够简化许多复杂的工作,特别是对于天然产物分子结构的解析.对于Solidagolactone (4) 结构的解析,能够为今后研究该物质分子结构对生物活性影响方面提供理论基础.
[1] 马腾, 翟晶, 白虹, 等. 加拿大一枝黄花化学成分的研究[J]. 食品与药品, 2011, 13(3): 104-106.
[2] 孙汉董. 二萜化学[M]. 北京: 化学工业出版社, 2012: 54-55.
[3] Müller D S, Untiedt N L, Dieskau A P, et al. Constructing quaternary stereogenic centers using tertiary organocuprates and tertiary radicals. Total synthesis of trans-clerodane natural products [J]. J Am Chem Soc, 2015, 137: 660-663.
[4] Enriz R D, Baldoni H A, Zamora M A, et al. Structure-antifeedant activity relationship of clerodane diterpenoids. comparative study with withanolides and azadirachtin [J]. J Agric Food Chem, 2000, 48: 1384-1392.
[5] Pichierri F. Molecular structure and conformations of caramboxin, a natural neurotoxin from the star fruit: a computational study [J]. J Mol Struct, 2015, 1079: 274-280.
Computation Study on Molecular Structure and Conformation of Solidago Lactone
ZHU Tianwen
(College of Chemical Science and Engineering, Yunnan University, Kunming, China 650500)
Solidago Lactone is a sort of Clerodane Diterpenoid natural products extracted from the solidago decurrens lour. It has antifeedant property to many insects. Firstly, this study builds 4 different kinds of spatial structures of the solidago lactone (4). Then, the energy value of all molecular conformations is calculated by means of DFT (Density Functional Theory) in order to discover thr relatively stable chemical shift value calculation from two conformations. The research shows that the branched chain conformation will have some influence on the molecular stability. Besides, there exist some errors between the chemical shift values and the experimental values. The correction factor can be introduced to modify the results.
Solidago Lactone ; Clerodane Diterpenoid; Molecular Conformation; Chemical Shift; Branched Chain
O6-04
A
1674-3563(2016)01-0036-06
10.3875/j.issn.1674-3563.2016.01.005 本文的PDF文件可以从xuebao.wzu.edu.cn获得
(编辑:封毅)
2015-06-12
朱天文(1993- ),男,浙江温州人,研究方向:天然产物的理论计算
猜你喜欢
同位素(2022年6期)2022-12-30
中草药(2022年1期)2022-01-13
昆明医科大学学报(2021年5期)2021-07-22
阅读与作文(小学低年级版)(2021年3期)2021-05-07
唐山师范学院学报(2020年6期)2020-04-16
唐山师范学院学报(2019年3期)2019-06-18
新青年(2018年3期)2018-03-16
小天使·六年级语数英综合(2017年6期)2017-06-06
北方音乐(2017年4期)2017-05-04
云南中医学院学报(2015年3期)2015-07-31
