
STAT3信号通路在神经退行性疾病中的研究进展
2024-03-13 07:48王珏陈佩弦何玲孙逸中国药科大学药学院南京211198
中南药学 2024年1期
王珏,陈佩弦,何玲,孙逸(中国药科大学药学院,南京 211198)
神经退行性疾病是以神经元进行性丢失和退行性病变为基础的慢性疾病,主要包括阿尔茨海默病(Alzheimer’s disease,AD)、帕金森病(Parkinson’s disease,PD)、血管性痴呆(vascular dementia,VaD)、亨廷顿病(Huntington’s disease,HD)、肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)、多发性硬化(multiple sclerosis,MS)等[1]。免疫系统中的多种细胞参与了神经退行性疾病的发展,包括小胶质细胞、星形胶质细胞和一些外周炎症细胞(例如巨噬细胞[2])。信号传导及转录激活蛋白3(signal transducer and activator of transcription 3,STAT3)是信号传导及转录激活蛋白(signal transducer and activator of transcription,STAT)家族的一员,是存在于神经元、血管内皮细胞、星形胶质细胞和小胶质细胞中的转录因子[3]。STAT3通过Janus激酶(Janus kinase,JAKs)磷酸化激活,响应上游信号(如细胞因子、生长因子等)的作用,进一步引起细胞增殖、分化和凋亡等[4]。近年来研究表明,STAT3信号通路与神经疾病病理状态下胶质细胞的激活和分裂密切相关,在神经退行性疾病的发生发展中发挥着重要作用[5-6]。本文就STAT3信号通路在神经退行性疾病中的作用及其潜在治疗意义进行综述。
1 STAT3信号通路
STAT3是STAT蛋白家族的一员,该家族迄今为止已经在人类基因组中确定了7 个STAT基因STAT1、STAT2、STAT3、STAT4、STAT5a、STAT5b和STAT6,在细胞增殖、分化、凋亡和血管生成等生理过程中起着复杂而重要的调节作用[7]。如图1所示,与其他家族成员相同,STAT3由一些在结构和功能上保守的区域组成,SH2 结构域(Src homology 2 domain,SH2 domain)和N端结构域(N-terminal domain,ND),在STAT3单体激活过程中介导二聚化,SH2 结构域高度保守,是大多数STAT3 抑制剂的靶点;螺旋线圈结构域(coiled-coil domain,CCD)通过蛋白- 蛋白相互作用作为核定位信号,而C 端转录激活结构域(C-terminal transcriptional activation domain,AD) 经历丝氨酸磷酸化,从而招募更多的转录激活因子,增强STAT3 的转录活性;DNA 结合域(DNA-binding domain,DBD),决定DNA 的结合;另外有连接子结构域(linker domain,LK)负责DBD 和SH2结构域的连接[8]。
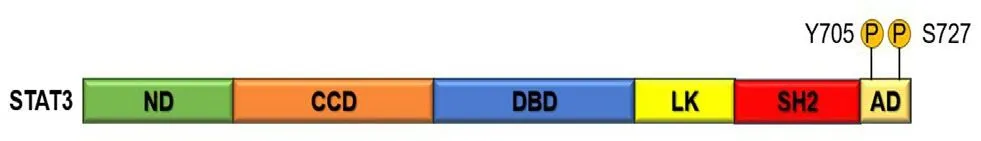
图1 STAT3的组成Fig 1 Structure of STAT3
JAK/STAT(Janus kinase/signal transducer and activator of transcription,JAK/STAT)信号通路介导STAT3的激活过程[9]。JAK/STAT级联由三个主要成分组成:一个细胞表面受体,一个Janus激酶(JAKs)和两个STATs蛋白[9]。如图2所示,当细胞因子与其受体结合时,JAKs被激活并传递调节信号;激活的JAKs使受体酪氨酸残基磷酸化,为具有SH2结构域的蛋白质创造结合位点;随即招募包含SH2结构域的STAT3,其酪氨酸残基被JAKs磷酸化并激活;这些激活的STAT3形成二聚体,并转移到细胞核,诱导靶基因的转录[10]。

图2 JAK/STAT信号通路Fig 2 JAK/STAT signaling pathway
STAT3的调控复杂而精密,主要通过磷酸化、乙酰化、甲基化、泛素化和酪氨酸磷酸化等翻译后修饰进行[11]。从图2可知,酪氨酸残基的磷酸化是STAT3的激活标志,一些激酶与STAT3磷酸化有关,包括p38蛋白、细胞外信号调节蛋白激酶、JUN的N端激酶和蛋白激酶Cδ等[12]。此外,JAK/STAT3信号通路也受到不同机制负调节因子的调节,例如细胞因子信号传导抑制(suppressors of cytokine signaling,SOCS)蛋白,作为经典的负反馈因子,可使JAKs失活并阻止STAT3进入受体结合位点[13]。此外,STAT家族成员间的交叉调控对细胞因子信号有着关键影响,STATs形成同源或异源二聚体,产生三种可能的DNA结合活性,包括STAT1-STAT1、STAT3-STAT3和STAT1-STAT3[14]。
STAT3通过改变效应细胞中的基因转录,响应细胞外信号分子的作用[15]。然而,在STAT3参与的通路中可能发生许多异常现象,导致STAT3调控异常,引发各种病理事件,如癌症、免疫缺陷综合征、炎症和神经退行性疾病等[16]。目前,关于STAT3的探索和研究主要集中在肿瘤病理领域,其在肿瘤细胞的免疫逃逸、扩散和转移方面发挥了至关重要的作用[8]。但近年来,多项研究证明,STAT3与神经元凋亡、神经炎症的表达及氧化应激的发生密切相关,在神经疾病领域同样有着不容忽视的作用[17]。
2 STAT3信号通路在神经退行性疾病中的作用
STAT3在中枢神经系统(central nervous system,CNS)中扮演着重要角色,不仅参与神经发育的多个生理过程,还在神经细胞存活和再生中起到关键作用[18-20]。值得注意的是,STAT3与神经胶质细胞的分化、分裂以及激活密切相关,而这类细胞对神经功能的影响巨大[21]。星形胶质细胞作为人类CNS中含量最为丰富的细胞,能够对CNS中的各种损伤作出反应从而激活;小胶质细胞作为神经系统的固有免疫细胞,是CNS的第一道防线,其在细菌感染或CNS损伤时迅速激活并释放多种细胞因子,并且小胶质细胞也被证明能参与到星形胶质细胞反应性增生中[22-23]。已有研究证实,在AD、癫痫和脑缺血等多种神经疾病的患者体内观察到STAT3的激活,同时伴有严重的神经炎症和氧化应激反应[24-26]。STAT3信号通路与神经退行性疾病的发生发展之间存在复杂而密切的联系,提示干预STAT3可能成为治疗该类疾病潜在的新策略。STAT3信号通路在神经退行性疾病中的研究尚属新兴领域,但其研究前景不容忽视。本文将对STAT3信号通路在神经退行性疾病中的研究加以综述。
2.1 AD
AD是一种典型的神经退行性疾病,表现为认知和记忆功能障碍[27]。AD在临床上难以被逆转,现有的药物仅能延缓病情发展[28]。AD 最显著的病理特征是神经纤维缠结以及老年斑产生,因此衍生出相应的两种假说:β-淀粉样蛋白(amyloidβ-protein,Aβ)的毒性假说和Tau(microtubule-associated protein tau,Tau)蛋白过度磷酸化假说[29]。但以上两种假说与AD发生发展之间的因果关系仍未得到完全证实[30-31]。面对AD研究的难关,我们应寻求更新的解决办法。
AD患者的早期特征是神经元内Aβ的积累,进而引起斑块周围的星形胶质细胞发生增生[32]。体内外实验证明Aβ与STAT3激活有关。在海马注射Aβ诱导的AD模型小鼠及APP/PS1(amyloid beta precursor protein/ presenilin 1,APP/PS1)转基因小鼠、5×FAD转基因小鼠的皮层和海马中均观察到STAT3 的酪氨酸磷酸化升高[33]。体外用Aβ刺激原代培养的大鼠神经元或胶质细胞也会诱导STAT3 磷酸化水平升高[33-34],说明抑制STAT3有可能改善AD病理损伤。在星形胶质细胞中通过siRNA干扰Stat3的表达可以减少白细胞介素-1β(interleukin-1β,IL-1β)和肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α),抑制星形胶质细胞激活,并部分缓解神经元损伤[35]。在体实验表明,在星形胶质细胞中特异性敲除Stat3下调了促炎因子,恢复了轴突功能,改善了老年APP/PS1小鼠的空间学习记忆能力[36]。有研究表明,在另一种AD模型动物5×E4小鼠中抑制STAT3可降低β-分泌酶1(β-secretase 1,BACE1)活性及上调脑内低密度脂蛋白受体相关蛋白1(low density lipoprotein receptor related protein 1,LRP-1)水平,从而减少大脑淀粉样蛋白斑块,减轻神经炎症和氧化应激反应,改善脑血管功能和脑血流动力学,最终提高小鼠的认知功能[24]。由此表明STAT3是星形胶质细胞增生相关通路中的关键调节因子,与该细胞介导的神经元存活、轴突再生和突触恢复密切相关[37]。综上,STAT3可通过调节神经元及异常激活的胶质细胞在AD的发生发展中发挥广泛而多样的作用,抑制STAT3有助于Aβ的清除,有望成为AD治疗的新方向。
作为AD的另一重要特征,Tau病理是指神经系统中过度磷酸化的Tau蛋白错误折叠并蓄积[38]。补体3a受体(complement C3a receptor,C3aR)是调节中枢免疫动态平衡的关键因子,而P-STAT3是C3aR的直接下游靶点[39]。Alexandra等[40]在PS19小鼠的原代小胶质细胞中敲除C3ar1基因或抑制STAT3磷酸化都能减少Tau蛋白蓄积,改善突触损伤和神经元丢失[39]。然而,关于STAT3与磷酸化Tau蛋白之间的关系尚存在不同观点。有体内研究发现,在P301L(frontotem-poral dementia with Parkinsonism-17 mutation,P301L)转基因小鼠中过表达的全长人Tau蛋白(full-length human tau40,hTau)通过增加乙酰化STAT1 和胞质中STAT3的相互作用,抑制STAT3从细胞质转运到细胞核,导致STAT3失活,而STAT3失活会进一步造成自噬障碍,引起可溶性及不溶性Tau蛋白的增加,从而恶化认知障碍[41]。这说明STAT3在Tau病理中发挥的作用是复杂的,有待进一步研究。
除了Aβ的毒性假说和Tau蛋白过度磷酸化假说,载脂蛋白E基因(apolipoprotein-E,Apoe)的ε4等位基因是AD最重要的遗传诱因[42]。一项蛋白质组学研究发现,相较于非携带者和老年AD患者,携带Apoe基因的年轻患者(没有显著AD症状)体内存在STAT3的激活,同时体外研究发现抑制STAT3缓解了神经炎症、减少了Tau蛋白磷酸化和Aβ1-42的积累,这提示STAT3或许是联系AD和Apoe基因的关键分子,为遗传性AD发病机制的研究提供了新的方向[43]。以上研究表明,STAT3信号通路与AD的发生发展密切相关,可能成为治疗AD 的潜在靶点,但后续还需要更多的临床前及临床证据证实。
2.2 PD
PD是一种常见的神经退行性疾病,主要表现为多巴胺能黑质-纹状体系统受损导致的运动功能障碍[44]。更具体地说,由于从黑质致密部投射到纹状体尾壳核的多巴胺能神经元异常死亡,导致多巴胺神经传递的丧失,机体出现各种运动症状,包括静止性震颤、运动迟缓、肌强直和姿势步态障碍。尽管PD最初被描述为一种没有痴呆的运动障碍疾病,但现在普遍认为PD的进展会影响其他黑质外多巴胺能、胆碱能和血清素能神经,导致包括嗅觉丧失、睡眠障碍和便秘在内的非运动症状,以及痴呆和抑郁症等认知和精神症状[45]。
PD中聚集的α-突触核蛋白会使小胶质细胞向促炎表型(M1)转化,从而加速疾病进程[46]。有研究者通过定量蛋白质组学光谱分析法,发现重组α-突触核蛋白刺激的小胶质细胞及脑内注射α-突触核蛋白预制纤维的小鼠中均存在STAT3通路激活[47],说明STAT3参与小胶质细胞的活化及PD的发病过程。还有体外研究表明M1型小胶质细胞释放的过氧化氢能诱导原代星形胶质细胞中STAT3的激活,使得胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)的表达升高,导致反应性星形胶质细胞增生,进一步损伤多巴胺能黑质-纹状体系统,导致运动障碍[48]。由于在大鼠和人GFAP启动子中都发现了潜在的STAT3结合位点[49-50],故STAT3可能通过诱导编码GFAP的Gfa基因表达上调从而介导星形胶质细胞活化,影响PD的疾病发展。钙离子是细胞功能的重要次级信使,储存在内质网和线粒体中,而钙稳态失调是PD发病机制的另一个关键特征[51]。Bo等[52]在小胶质细胞中敲低Calhm2(calcium homeostasis modulator 2,Calhm2)或EFhd2(EF-hand domain family member D2,EFhd2)均可显著抑制细菌脂多糖(lipopolysaccharide,LPS)诱导的STAT3激活,抑制炎症因子释放,保护神经元。这表明STAT3功能与钙稳态失调息息相关,调控STAT3可通过钙稳态参与PD的病理过程。然而关于STAT3在PD发生发展中的作用也存在争议。有研究证明在1-甲基-4-苯基吡啶离子(1-methyl-4-phenyl-pyridine,MPP+)处理的星形胶质细胞模型和1-甲基-4-苯基-1,2,3,6-四氢吡啶(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,MPTP)诱导的PD小鼠中激活星形胶质细胞的IL-6/JAK/STAT3通路,可上调谷氨酸转运蛋白(glutamate transporter,GLT),阻止MPP+/ MPTP诱导的谷氨酸再摄取能力的降低及脑内谷氨酸能系统的过度激活,有利于改善PD中多巴胺能神经元的退化[53]。
综上,STAT3与PD的多个病理特征具有不容忽视的相关性,但与PD发病机制的关系仍不明确,大多数研究集中于体外,有待进一步探索。
2.3 VaD
VaD是一种由脑血管疾病引起的认知功能障碍综合征,由出血或缺血造成的急慢性脑缺氧是其发生发展过程中的高危诱因[54]。目前,VaD的发病机制尚未完全阐明,但多项体内外研究证实神经炎症是VaD发生的重要机制之一,而STAT3是VaD病理过程中重要的炎症诱导因子[55-56]。血管性认知障碍患者在VaD的临床前、临床和严重阶段均存在广泛慢性炎症反应[57-59]。慢性白质缺血性损伤引起的VaD模型动物脑内STAT3表达水平和磷酸化水平的升高,小胶质细胞向M1表型极化,胼胝体脱髓鞘和Ranvier结节紊乱,工作记忆表现下降[60]。除了人体和动物模型,在体外以低氧刺激诱导海马神经元细胞损伤模拟VaD的病理环境时,也观察到STAT3磷酸化水平升高,促炎因子增多,内质网应激增强[61]。由此可见,STAT3与VaD的发生发展密切相关,抑制STAT3可能改善VaD。在双侧颈总动脉闭塞诱导的VaD模型大鼠中,采用电针刺激可下调海马和杏仁核中STAT3的表达水平,可降低神经炎症反应,改善模型动物的学习和记忆能力[62]。此外,一些中药制剂或天然产物改善VaD的作用也与STAT3有关。清脑益智汤可抑制体外培养的原代神经元中STAT3的表达及磷酸化水平,抑制胶质细胞的活化[63]。脑保健颗粒可通过抑制JAK2/STAT3通路,减少VaD模型大鼠促炎细胞因子的产生,并进一步降低神经炎症反应,从而保护缺血性脑损伤,显著缓解VaD症状[64]。经典的汤剂加味二陈汤可通过JAK2/STAT3通路降低VaD大鼠脑内Aβ1-42的水平,提高抗氧化能力,并降低炎症因子水平,增加神经元的数量,改善认知功能障碍[65]。木兰叶中分离出来的天然化合物厚朴酚在体内和体外实验中均可以通过靶向沉默调节蛋白-3(sirtuin 3,SIRT3)来抑制转录因子STAT3的磷酸化和核易位,减少星形胶质细胞A1型的极化,改善神经炎症和突触损伤,从而缓解慢性脑灌注不足诱导的VaD症状[66]。因此,STAT3可能是治疗VaD的靶点,抑制STAT3信号通路可以有效减少神经炎症损伤并改善症状,但目前还缺乏关于临床应用的安全性和有效性数据,未来还需进一步的深入研究。
2.4 HD
HD是一种常染色体显性遗传的神经退行性疾病,其特征是认知、运动和精神障碍[67]。HD的主要病因是亨廷顿基因中显性遗传的CAG三核苷酸重复扩增,表达出了异常的亨廷顿蛋白,该蛋白聚集后形成大分子团,最终在脑中积聚;在细胞水平上,异常的亨廷顿蛋白通过多种机制导致神经元功能障碍和死亡,包括破坏蛋白质内稳态、蛋白转录、线粒体功能,随着病情恶化,纹状体早期可见表观变化并累及皮层[68]。在HD的诸多发病机制中,氧化应激和炎症的级联反应是使HD恶化的两个重要因素[69]。HD患者和许多模型动物均表现出炎症反应及STAT3激活。有研究者通过流式细胞术在HD患者离体的外周单核细胞中发现促炎细胞因子IL-6水平升高,JAK/STAT信号通路激活[70]。还有研究者发现HD患者的壳核中STAT3表达显著升高[71]。在小鼠和非人类灵长类动物HD遗传模型的反应性星形胶质细胞中也存在STAT3激活[5]。对野生型小鼠双侧纹状体注射能表达突变亨廷顿蛋白的腺相关病毒,小鼠的神经元、小胶质细胞和星形胶质细胞中均出现STAT3表达量和磷酸化水平升高,小鼠也表现出明显的运动能力障碍[72]。这说明STAT3信号通路在神经炎症中发挥重要作用,通过抑制STAT3介导的炎症反应通路可能可以作为治疗HD的潜在新疗法。Jang等[73]研究显示,一种从五味子根中分离出来的去甲三萜类化合物五味子内酯C在体内和体外模型中都表现出通过抑制小胶质细胞的STAT3信号通路发挥缓解行为功能障碍、纹状体变性和减少神经炎症反应的作用。但是与抑制STAT3信号通路降低神经炎症而发挥神经元保护作用不同,其他研究者还发现激活STAT3信号通路可以改善HD患者病理特征。HD发病与衰老密切相关的原因在于患者体内miR-29b-3p随着年龄逐渐增多,miR-29b-3p可通过抑制STAT3抑制自噬,使突变的亨廷顿蛋白聚集,而进一步损伤纹状体中型棘神经元(medium spiny neuron,MSN),说明维持STAT3的正常活性对延缓HD的发生和疾病进展具有积极意义[74]。在过表达人亨廷顿蛋白的纹状体祖细胞系中上调STAT3信号通路可诱导碱性螺旋-环-螺旋转录因子1(twist1)的表达,减少脑细胞凋亡和损伤,进而对HD患者纹状体的变性产生保护作用[75]。Abjean等[71]研究发现在HD小鼠的星形胶质细胞中激活JAK2-STAT3信号通路可以增强溶酶体和泛素蛋白酶体降解系统的蛋白水解能力,减少突变亨廷顿蛋白的聚集,改善HD小鼠的神经缺陷[71]。重组人促红细胞生成素(recombinant human erythropoietin,rhEPO)和干扰素-β-1b(interferon-β-1b,IFN-β-1b)也可通过激活JAK1/STAT3通路显著改善3-硝基丙酸诱导的HD大鼠的运动和行为能力,恢复线粒体功能以及脑源性神经营养因子水平,减少纹状体中的氧化应激和炎症反应[76]。由此可见,STAT3的作用机制十分复杂,STAT3信号通路在HD中的作用仍存在争议,可能在反应性星形胶质细胞和神经元之间存在双向作用,还需进一步的研究。
2.5 ALS
ALS是一种以脑和脊髓中运动神经元进行性损失为特征的神经退行性疾病,表现为进行性运动无力和神经源性肌萎缩,目前缺乏有效的治疗药物[77]。有研究发现,ALS患者存在轴突病变和神经炎症,其脊髓内神经元和胶质细胞中STAT3表达增多,肌肉组织中STAT3和促炎细胞因子IL-6表达都上调[78-79]。有研究对ALS患者的成纤维细胞进行原代培养,发现细胞中的STAT3也持续上调[80]。此外,一种ALS小鼠模型——SOD1G93A转基因小鼠的脊髓运动神经元、反应性星形胶质细胞和激活的小胶质细胞中也存在STAT3的磷酸化活化和核转位水平升高,并进一步诱导多种促炎基因产物表达上调[81]。同样,在另一种家族性ALS的遗传模型动物的脊髓α-运动神经元中,STAT3在轴突损伤、缺血和过量谷氨酸等多种刺激下可被激活[82]。这些发现说明,STAT3在ALS患者和动物模型中会发生不同程度的激活,STAT3信号介导的炎症反应可能与ALS的发病机制有关,STAT3信号通路可能是阐明ALS病理过程的关键。TDP-43(TAR DNA binding protein-43,TDP-43)是由Tardbp基因编码的核内蛋白,在ALS患者神经元细胞质中以异常的不溶性聚集物形式积累,是ALS重要的病理特征[83]。而STAT3抑制剂氯硝柳胺可以阻止TDP-43由细胞核向细胞质的错误转移并降解胞质中的TDP-43聚集体,进而减弱应激状态下神经元的形态变化,这一发现为ALS的发病机制研究及治疗提供了新的思路[84]。上述研究结果表明,STAT3信号通路与ALS发病机制密切相关,研究STAT3在ALS发生发展中的作用可能对阐明ALS的发病机制并发现新的药物靶点具有重要意义。
2.6 MS
MS是一种慢性神经系统疾病,以中枢神经系统炎症和脱髓鞘为主要特征,并伴有不同程度的轴突和神经元损伤[85]。近几年的研究发现,STAT3信号通路与MS的病理过程密切相关[86]。蛋白肽PLP139-151诱导的实验性变态反应性脑脊髓炎(experimental autoimmune encephalomyelitis,EAE)模型小鼠中的脾脏和脑内及酮腙诱导的脱髓鞘小鼠的脑组织中均存在STAT3信号通路的激活[87-88]。此外,STAT3信号通路在髓系细胞和淋巴系细胞中也发挥关键作用,通过对该通路的阻断可减弱神经元的炎症反应,从而改善MS疾病症状,这说明抑制STAT3可能是治疗MS的一种潜在策略[89]。目前已有报道,一些小分子化合物、天然产物可通过抑制STAT3介导的炎症反应来改善MS的相关病理症状。葫芦素B可以阻断STAT3的磷酸化过程,在神经炎症和自身免疫性疾病中发挥免疫调节作用,减少MS早期阶段的神经元缺陷[90]。古古甾醇是一种来自CommiphoraWightii树的植物甾醇,具有强大的抗炎作用,而且可以通过调控JAK/STAT信号通路来改善与较低STAT3水平相关的行为缺陷,提高大鼠脑内过氧化物酶体增殖物激活受体-γ(peroxisome proliferator activated receptor-γ,PPAR-γ)和髓鞘碱性蛋白的水平,保护神经[91-92]。而新型药物β-D-甘露糖醛酸(β-D-mannuronic acid,M2000)可通过下调继发型MS患者中的白细胞介素-17(interleukin-17,IL-17)、STAT1和STAT3的相关基因,并降低外周血单核细胞中TLR2(Toll like receptor 2,TLR2)和TLR4(Toll like receptor 4,TLR4)的表达,从而降低炎症反应[93]。芬戈莫德(fingolimod,FTY720)最初被证明是一种1-磷酸鞘氨醇受体的调节剂,可通过抑制自身免疫反应治疗MS。后来发现芬戈莫德通过调节STAT3信号通路减弱脑白质缺血后小胶质细胞产生的神经炎症,进而使小胶质细胞向抗炎状态转化,提示芬戈莫德可用于治疗其他神经炎症为主要病理特征的疾病[60]。在MS的诸多发病机制中,免疫反应也是推动疾病复发和进展的重要因素,而STAT3也可通过调节免疫反应缓解疾病[94]。Aqel等[95]研究发现新型小分子前药LLL12b通过特异性抑制STAT3进而重建Teff(effector T cell,Teff)与Treg(regulatory T cell,Treg)的平衡,可以阻止致病的Teff细胞增多,避免进一步的炎症反应。上述研究结果提示STAT3信号通路具有作为MS靶点的潜力,抑制STAT3介导的炎症反应为治疗MS开辟了新的维度,并为阐明MS的病理机制提供新的角度。
3 STAT3通路相关调控剂
在大多神经退行性疾病中,抑制STAT3能改善胶质细胞的过度激活,减轻神经炎症和氧化应激。因此,寻找合适的STAT3抑制剂或许是可行的治疗手段。
3.1 靶向STAT3的小分子抑制剂
目前关于靶向STAT3的小分子抑制剂的研究较多,但大多集中于肿瘤领域,在神经退行性疾病中的研究还十分有限。大多数小分子抑制剂可以选择性地结合到STAT3的DBD、SH2结构域,从而抑制其表达或激活。然而,大部分小分子抑制剂由于水溶性和细胞渗透性较低,临床预期较差[10]。BBI608(napabucasin)是迄今为止唯一进入Ⅲ期临床试验的STAT3抑制剂,通过选择性结合DBD结构域发挥抑制STAT3的作用,用于晚期肿瘤治疗。该抑制剂在临床使用中最常见的不良反应是腹泻、恶心、呕吐、腹部痉挛和疲劳[96]。
氯硝柳胺是FDA批准的抗绦虫药物[97],是STAT3抑制剂,其通过抑制STAT3,阻止了运动神经元内TDP-43(TAR DNA binding protein-43,TDP-43)从胞核向胞浆的转移,并降解TDP-43聚集体,减少了应激条件下细胞线粒体三磷酸腺苷(ATP)的产生,氧化应激反应和细胞凋亡,并缓解了细胞损伤,从而改善了ALS的病理特征[84]。一项随机、双盲的临床研究显示,与安慰剂组相比,除轻微的刺激反应外,未见显著的不良反应[98]。但是与大多数小分子STAT3抑制剂相似,氯硝柳胺的水溶性有待进一步改善[99]。有研究者在氯硝柳胺中引入了亲水基团得到化合物NGT 02,水溶性和生物利用度得到改善,有效地诱导肿瘤细胞的凋亡并抑制癌细胞的迁移[99]。笔者前期也研究了氯硝柳胺对AD小鼠的作用,发现Aβ1-42诱导的AD小鼠给予氯硝柳胺后,小鼠脑内神经炎症和氧化应激反应均降低,学习记忆能力提高,这提示了氯硝柳胺在治疗认知障碍相关疾病中具有潜在价值;研究还表明氯硝柳胺在25 mg·kg-1剂量下连续给药7 d对小鼠的心、肝、肺、脾、肾等主要器官的正常组织结构无明显影响,说明该给药策略安全有效[100]。羟氯喹也是一种FDA批准的抗风湿药物,已被证明是一种能穿透血脑屏障的STAT3抑制剂。动物实验结果显示APP/PS1小鼠给予羟氯喹后海马突触可塑性和认知功能均得到改善[101],说明STAT3是老药新用的潜在靶点。一项荟萃分析显示,与安慰剂组相比,羟氯喹临床研究中最为显著的不良反应是皮肤色素沉积,其次是视网膜病变[102]。
一些处于临床前研究阶段的人工合成的小分子化合物也可通过抑制STAT3改善认知功能。LLL-12是根据姜黄素的结构合成的小分子STAT3抑制剂,能直接抑制STAT3的705位酪氨酸磷酸化并阻止其激活,可减少5×E4小鼠大脑淀粉样斑块沉积并改善其认知功能[24]。C188-9是计算机辅助合成的磺胺类化合物,可选择性地与STAT3中SH2结构域结合,抑制STAT3激活的过程[103]。在BV2细胞的炎症模型和Aβ1-42清除实验中使用该抑制剂可下调促炎因子、降低Aβ1-42的水平,并且在SH-SY5Y-hTau441细胞中能减少磷酸化Tau蛋白的水平[43]。
3.2 抑制STAT3的天然产物
除了小分子抑制剂,在自然界中存在具有抑制STAT3活性的天然化合物,它们大多药理作用复杂且作用广泛,对其作用机制的研究有助于开发结构新颖、活性更强的新药。
染料木素是一种从大豆提取物中提取的异黄酮。它是一种蛋白酪氨酸酶抑制剂,具有强烈的抗血管生成、抗自由基、抗氧化、抗炎症和抗肿瘤作用,可通过抑制JAK2/STAT3通路以及激活KEAP1/NRF2通路,抑制小胶质细胞和星形胶质细胞的激活,减轻癫痫发作的强度和持续时间,增加抗凋亡蛋白和神经元存活[104]。三七总皂苷是血栓通注射液的主要成分,可通过抑制JNK介导的JAK2/STAT3和NF-κB信号通路,缓解炎症反应,从而改善脑缺血再灌注大鼠脑微血管结构的损伤和功能障碍[105]。
相关靶向STAT3通路的小分子抑制剂及抑制STAT3的天然产物总结见表1。

表1 STAT3抑制剂Tab 1 STAT3 inhibitors
4 总结与展望
神经退行性疾病是目前困扰患者的广发慢性疾病,难以治愈且症状严重,已经引起了社会广泛的关注。然而其发病机制研究尚不全面。本文总结了STAT3信号通路在CNS中的相关机制研究,该通路是神经胶质细胞激活的关键诱因,与神经退行性疾病中常见的神经炎症、病理性蛋白聚集、突触和神经网络障碍、细胞骨架异常、蛋白质内稳态异常、能量稳态失调等特征都有着潜在的联系。进一步阐明STAT3 通路在神经退行性疾病中的重要作用,对于理解疾病的发病机制至关重要,并且可能为神经退行性疾病的治疗提供潜在新靶点。
猜你喜欢
自然杂志(2021年6期)2021-12-23
神经损伤与功能重建(2020年11期)2020-12-01
天津医科大学学报(2019年6期)2019-08-13
现代装饰(2018年5期)2018-05-26
分析化学(2017年12期)2017-12-25
湖南中医药大学学报(2016年1期)2016-12-01
磁共振成像(2015年1期)2015-12-23
安徽医科大学学报(2015年9期)2015-12-16
电源技术(2015年5期)2015-08-22
弹箭与制导学报(2015年1期)2015-03-11
