
含氮有机液体储放氢催化体系研究进展
2024-01-16 11:35李佳豪杨锦潘伦钟勇斌王志敏王锦生张香文邹吉军
化工进展 2023年12期
李佳豪,杨锦,潘伦,钟勇斌,王志敏,王锦生,张香文,邹吉军
(1 天津大学化工学院,绿色合成与转化教育部重点实验室,天津 300072;2 东方电气集团东方锅炉股份有限公司,四川 成都 610000)
国家发展和改革委员会印发的《氢能产业发展中长期规划(2021—2035 年)》中指出,来源广泛、绿色低碳的氢能对构建清洁低碳、安全高效的能源体系以及实现碳达峰、碳中和的目标具有重要作用[1]。当前可再生能源,如风电、光伏发电发展迅速,可再生能源发电量占比正在持续增加,但由于其不连续性和不稳定性对电网的安全有效运行带来了一定的挑战[2]。未来可持续能源基础设施需要大规模能源储存以应对可再生能源(如太阳能、风能)的波动性,并促进从能量富集地区到能量贫乏地区的运输[3]。氢气是高效的储能载体,其储能具有长周期、大规模、不受自然环境和地域约束等优势,通过将可再生能源储存于氢能中可实现其大规模跨时、跨地应用[4]。氢气的开发及利用包括制氢(化石燃料制氢、电解水制氢等)、储运氢、终端用氢(如工业用氢、燃料电池)环节[5]。在制氢领域,除了传统的化石燃料制氢,中国石油、中国石化、国家能源集团、国家电网等多家央企率先投资绿氢项目建设[6-7];在用氢方面,除了在工业炼化上的应用,氢气在燃料电池方面也有示范应用[6]。而氢储运是连接上游制氢、下游用氢的关键环节,同样也是调节绿氢跨时空配置、灵活供应的重要保障[8]。
目前常用的储氢方法有高压压缩储氢[9][储氢罐压力一般为35~70MPa,储氢密度1%~3%(质量分数,下同)][10-12]、低温液化储氢[13](储存温度为-253℃,储氢密度10%左右[12,14])、有机液体储氢化合物(liquid organic hydrogen carriers,LOHCs)储氢(常温常压运输,储氢密度5%~10%)[15-16]、固体材料储氢(包括多孔材料物理吸附类,如沸石、碳纳米管,和金属氢化物如MgH2、LiBH4等,储氢密度7.6%~18.5%,但此类材料脱氢温度较高)[17]。其中,LOHCs储放氢载体在常温下多为密度较高的液体,可通过罐车、管道等方式完成运输,因而这种储氢方式备受关注,因其在储存条件上的适宜性,LOHCs 储氢是氢气储运技术的可选方案之一[18-19]。LOHCs 的储氢、放氢示意图如图1所示。首先对贫氢LOHCs 进行催化加氢,将氢气储存于有机液体化合物中,利用现有设施(罐车或管道等)运输至加氢站等氢气使用地,然后对富氢LOHCs 进行催化脱氢,释放出氢气并得到贫氢LOHCs,最后将贫氢LOHCs 运输回储氢所在地,实现氢气循环储放过程[20]。以LOHCs为载体可实现经济、安全、大规模和长周期的氢气储存[21],而且储氢密度较高(满足美国能源部车载储氢密度5.5%的技术要求[22]),在常温常压下能够使用现有液体燃料基础设施或罐车进行长距离运输[23-24]。
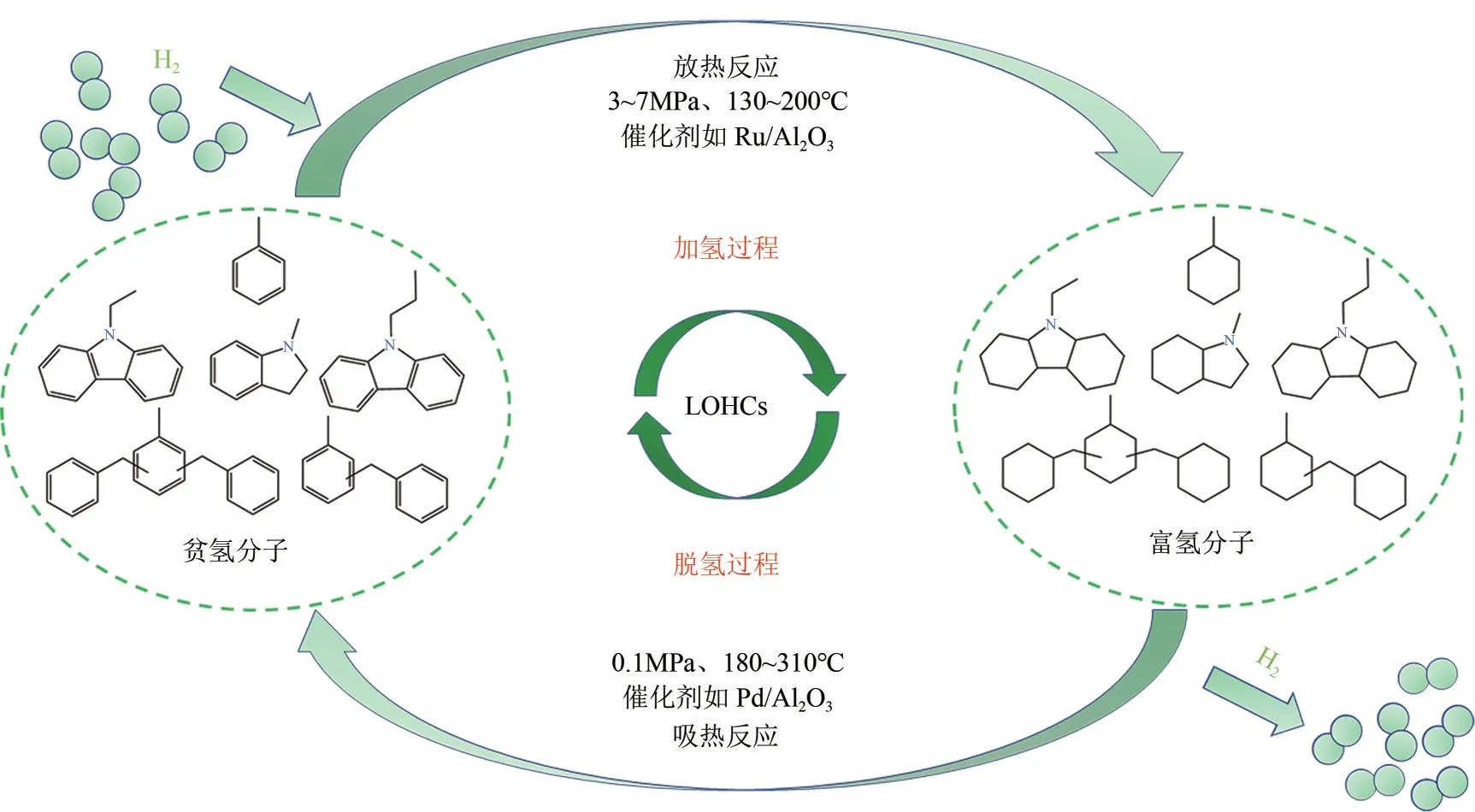
图1 基于LOHCs的氢气储存和释放循环过程示意图
目前报道的LOHCs 化合物从分子结构上可分为芳烃类和含氮杂环类[5],其中前者包括苯/环己烷[25]、甲苯/甲基环己烷[26-29]、二苄基甲苯(DBT)/十八氢二苄基甲苯(18H-DBT)[4,30-33]、单苄基甲苯(BT)/十二氢单苄基甲苯(12H-BT)[34-39]等;而后者包括N-乙基咔唑(NEC)/十二氢-N-乙基咔唑(12H-NEC)[25,40-43]、N-丙基咔唑(NPC)/十二氢-N-丙基咔唑(12H-NPC)[25,44]、吲哚/8H-吲哚[45-46]等。
含氮LOHCs体系中氮原子的引入降低了富氢分子脱氢反应焓[11](芳烃类18H-DBT、甲基环己烷和含氮类12H-NEC 脱氢反应焓分别是65kJ/mol、68.3kJ/mol和50kJ/mol[47-49]),有利于在更低温度下完成脱氢反应,如18H-DBT 的脱氢温度为290~310℃[50-51],而12H-NEC在200℃以下便可完成脱氢,因此含氮LOHCs在储运氢能效方面更具优势。一些典型含氮LOHCs的转化及其理化性质如表1所示。
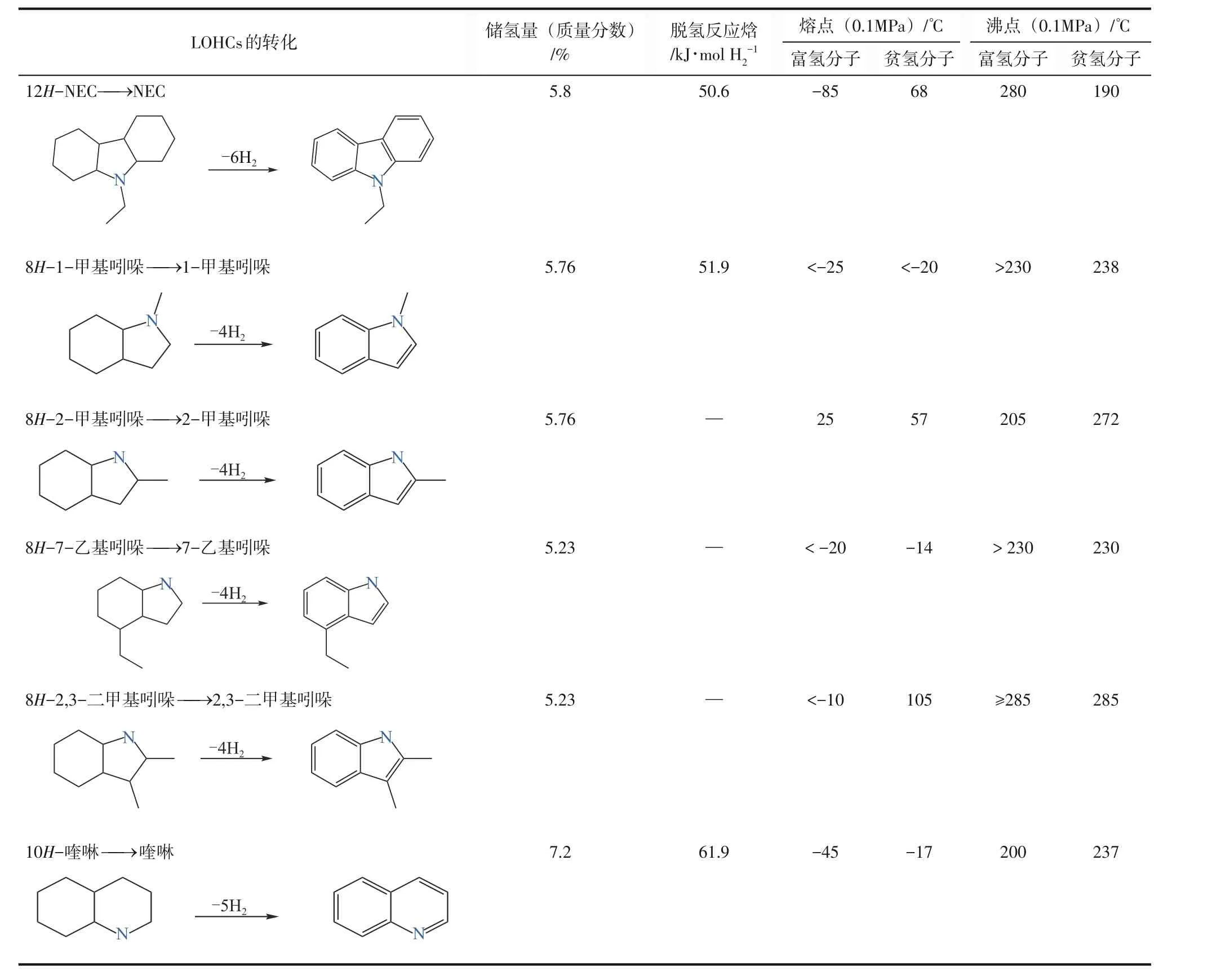
表1 典型的含氮LOHCs的转化及其理化性质[46,62-65]
尽管含氮LOHCs 受到广泛研究,但其中一些化合物的熔点较高[如2-甲基吲哚(2-MID)熔点为57℃、NEC 熔点为68℃][52],在常温下为固态,因此对催化反应及运输过程均带来一定困难。为了降低体系熔点,可采用全氢产物部分脱氢法,如将12H-NEC部分脱氢至4H-NEC,产物在常温下为液体[53],但是会降低1/3 的储氢密度;也可增加烷基咔唑中链烷基的长度,降低其熔点,但会造成储氢密度的降低;还可以采用不同烷基咔唑复配方法,如NEC-NPC-NBC(N-丁基咔唑)混合物的熔点可降低至13℃,储氢密度为5.5%,与NEC相当[52]。
目前已有一些文献对含氮LOHCs 的催化反应进行了概述[11,54-55],重点关注了加氢/脱氢催化剂结构设计,而缺少对该体系的反应机理、反应动力学、催化剂详细构效关系以及应用等方面的系统综述。因此,本文将全面介绍含氮LOHCs 体系的研究进展,包括加氢/脱氢过程的反应路径及催化机理、催化剂结构及反应条件对反应的影响规律,基于连串反应、反应网络等模型的动力学等,最后介绍该体系的相关应用并提出未来展望。
1 含氮LOHCs加氢/脱氢反应机理
1.1 加氢反应机理
1.1.1 反应路径
含氮LOHCs 加氢过程较为复杂,涉及多步反应和多个反应中间体,不同的平行加氢与异构反应步骤可能会同时发生,进而影响全氢产物的收率和分子立体构型[56]。解析其反应机理和反应路径对于催化剂选择和反应条件优化至关重要。以研究较多的NEC为例[57],其在催化加氢反应中(质量分数为5%的Ru/Al2O3)共产生4 种中间产物:4H-NEC、6H-NEC、8H-NEC 和10H-NEC。Sotoodeh 等[58]根据对液相产物分析提出了加氢反应路径并获得了各反应步骤的活化能,如图2(a)所示。对于NEC加氢的初始位置,理论计算表明,NEC 中两个芳环里最靠近N 原子的两个C 原子[图2(a)中C1、C2 或C11、C12]之间的π键会首先受到H自由基的攻击,因此NEC 的加氢过程从这两个碳原子之间的不饱和键开始[59]。NEC的转化包含两个平行反应:生成4H-NEC 和直接生成8H-NEC。而8H-NEC 的转化也遵循两条平行路径并分别生成10H-NEC和12HNEC。在NEC转换过程中,k1小于k2(与活化能结果相反),说明NEC 倾向于直接生成8H-NEC。在8H-NEC的转化过程中,k6远大于k5、k7,说明8HNEC可直接生成12H-NEC。
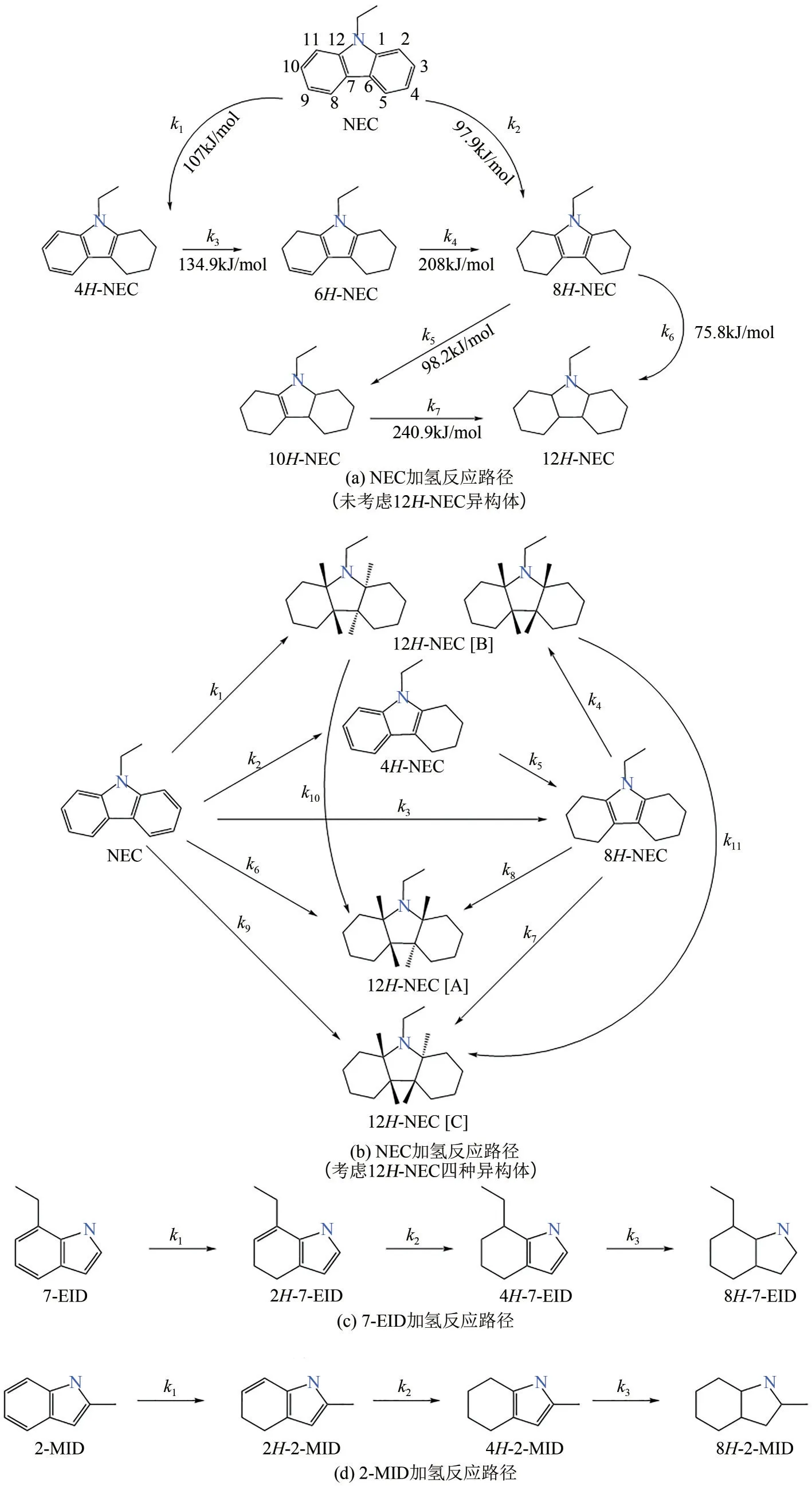
图2 含氮贫氢分子加氢反应路径[46,57-58,60-61]
另外,Eblagon等[60]在NEC的加氢反应路径中考虑了12H-NEC 的4 种同分异构体(12H-NEC [A]、12H-NEC [C]和两种12H-NEC [B]构型),如图2(b)所示。该加氢反应过程仅检测出4H-NEC 和8HNEC这两种中间产物,与上述机理一致。通过分析分子构型发现,完全加氢产物的主要构型为12HNEC [B](使用Ru基催化剂,12H-NEC [B]的选择性高达94%)。实验结果表明,k1~k5的值均远大于k6~k9,说明反应优势路径为NEC 经各中间体生成12H-NEC [B]。另外,全氢产物12H-NEC的各种构型分子之间也会相互转化,例如12H-NEC [B]会缓慢转化为12H-NEC [A]和12H-NEC [C]。
其他含氮LOHCs 加氢反应的路径主要为串联反应路径。Chen 等[46]用5% Ru/Al2O3催化剂对7-乙基吲哚(7-EID)加氢并提出串联加氢反应路径,如图2(c)所示。加氢过程首先从六元环开始,2H-7-EID 是主要中间产物,4H-7-EID 在整个反应过程中最高含量不到5%。此外,2-甲基吲哚(2-MID)的加氢反应路径[61]与7-EID 类似,如图2(d)所示。其中4H-2-MID为主要中间产物,反应过程中仅检测出微量2H-2-MID。
1.1.2 催化反应过程
在反应热力学方面,Eblagon 等[56]使用密度泛函理论(DFT)对NEC 加氢反应过程中96 种理论化合物进行建模以计算反应焓,并绘制了各种(中间)产物与反应物之间的能量差,结果如图3所示(纵坐标值负值越小表明化合物在气相中越稳定)。可以看出8H-NEC最左侧构型分子具有相比其他部分加氢中间体更低的能量差,表明其更稳定,是主要反应中间体,这与实验结果相一致。
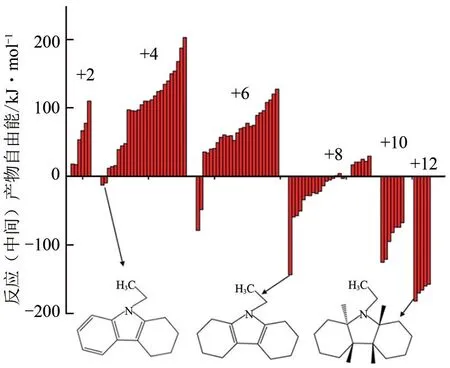
图3 气相中最稳定反应中间体/产物与NEC之间的自由能比较[56]
在反应动力学方面,具有平面多环结构的反应物和非平面结构中间体分子在不同活性位点上吸附强度有差异[66]。Eblagon 等[18]利用DFT 计算了NEC和8H-NEC 在Ru 表面的平台位点(Ru{001})和台阶位点(Ru{109})上的吸附。由图4(a)、(c)可知,NEC在Ru{001}晶面上的计算吸附能为-2.56eV,低于在Ru{109}上的吸附能,表明NEC 在Ru{001}晶面上吸附更稳定,这是由于平面芳环与平台位点具有更强的相互作用。图4(b)表明,动力学稳定的部分加氢产物8H-NEC不再是平面结构,新引入的H原子阻止了分子中其他物种(如N)与活性位表面的相互作用。若乙基指向平台平面,产生的额外空间位阻将进一步降低吸附强度。因此大部分中间体8H-NEC将脱附到溶液中,而不是在平台活性位继续反应形成最终产物12H-NEC。
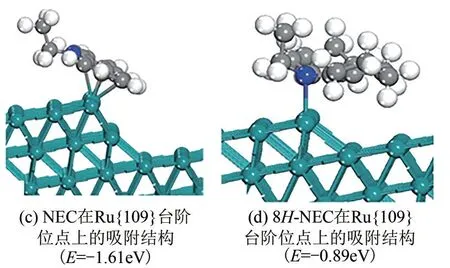
图4 NEC和8H-NEC在Ru{001}和Ru{109}位点上的吸附结构及其吸附能[18]
随后,在Ru{109}台阶位点上研究了中间产物8H-NEC 的吸附,结果如图4(d)所示。8H-NEC 在Ru{109}表面以N—Ru 键形式吸附,吸附能约为-0.89eV,明显低于8H-NEC 在平台位点的吸附能(-0.32eV)。因此,中间产物8H-NEC在台阶位点上吸附更稳定,这是由于分子吸附的空间位阻在台阶或边缘位点上被显著削弱。通过减小催化剂活性颗粒尺寸可以增加更多边缘和台阶位点,从而有利于8HNEC 的吸附并进一步促进加氢过程生成最终产物12H-NEC[66]。然而目前没有关于其他含氮LOHCs(如1-MID、2-MID、7-EID等)催化加氢机理的报道。
1.2 脱氢反应机理
1.2.1 反应路径
与加氢反应类似,含氮LOHCs 的脱氢反应也涉及多步过程。以最典型的12H-NEC 为例,其脱氢反应路径包括三步[57][图5(a)]:12H-NEC 首先脱去五元环中的H生成8H-NEC,然后脱去六元环中的H生成4H-NEC,最后脱去剩余H生成最终产物NEC。与之类似,全氢1-甲基吲哚(8H-1-MID)的脱氢也是从五元环开始[67],首先脱氢生成4H-1-MID,然后脱去六元环中的氢生成最终产物1-MID[图5(b)],全氢7-乙基吲哚(8H-7-EID)也表现出类似反应路径[图5(c)][46]。相比上述脱氢过程,全氢吲哚的脱氢反应路径较为复杂。Ouma等[68]采用DFT计算提出三条反应路径并计算出每步反应活化能,如图5(d)所示。反应路径2脱除6H的反应能垒明显低于路径1速控步骤(8H-吲哚至6H-吲哚),这表明路径2优于路径1。而路径3中8H-吲哚直接脱氢生成吲哚的能垒与路径2速控步骤相差不大,表明8H-吲哚的脱氢有可能同时按路径2 和路径3进行。
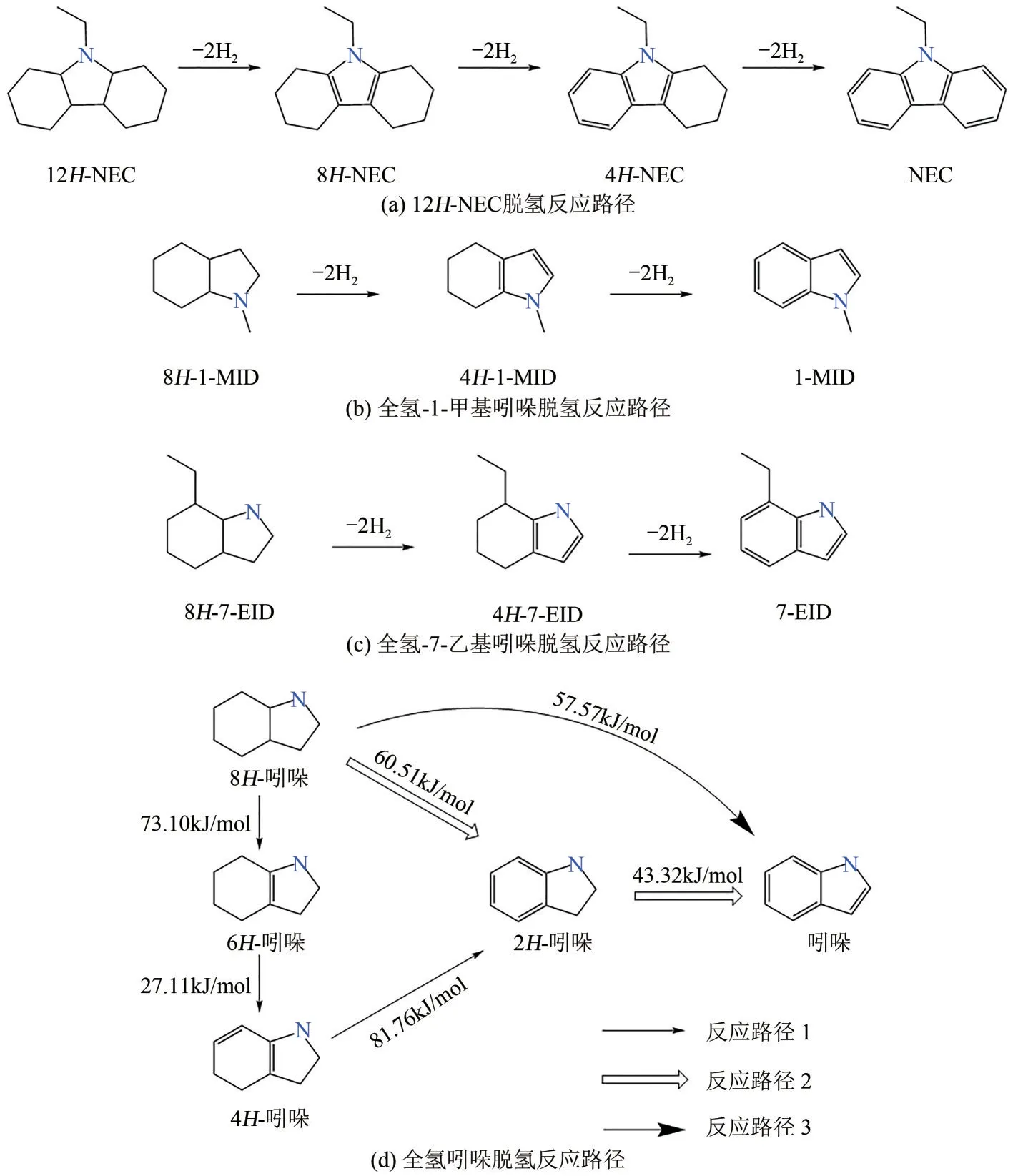
图5 含氮富氢分子脱氢反应路径[46,57,67-68]
1.2.2 催化反应过程
为了阐明12H-NEC 在负载型Pd催化剂上的脱氢机理,Sobota等[69]采用红外光谱(IRAS)和高分辨率X 射线光电子能谱(HR-XPS)分析了Pd/Al2O3/NiAl{110}催化剂表面的12H-NEC 脱氢过程。结果表明,在120K下,12H-NEC 分子吸附在活性位点Pd上。170K时,该分子的咔唑骨架C1或C12[图2(a)]处发生C—H键的断裂。温度超过350K时,饱和六元环的部分脱氢中间体在Pd 纳米颗粒上发生分子C—N键的断裂。500K以上,Pd纳米颗粒上的分子碎片进一步分解,产生小分子碎片(小分子烃类)和原子(C、H、N)。与之类似,Amende等[70-71]也发现,173~223K 下12H-NEC 吸附在Pd{110}面上,223K 时12H-NEC 发生C—H 键活化并快速脱氢生成稳定的中间体8H-NEC,273K 以上8H-NEC 中六元环进一步脱氢,而350K 以上就会发生副反应(脱烷基生成咔唑类物质)。对于其他含氮富氢分子的脱氢机理也有类似的研究,Schwarz 等[72]采用上述方法探究了Pd{111}晶面上2H-吲哚、8H-吲哚的脱氢过程。在330~390K 下,2H-吲哚的五元环脱氢并生成吲哚,而8H-吲哚脱氢过程中由于六元环的部分脱氢所以形成了与2H-吲哚不同的中间体。390K 以上所有物质均发生脱氢反应以及产生部分裂解副产物。
上述研究均是在高真空下对脱氢机理进行探究,另外也有研究采用理论计算来解析脱氢过程。Sotoodeh 等[73]利用DFT 计算了12H-NEC、8HNEC 和4H-NEC 在Pd{111}表面吸附的过程(吸附能 分 别 为-95kJ/mol、-56kJ/mol、-117.5kJ/mol),如图6(a)~(c)所示。12H-NEC 通过五元环中的两个H原子吸附在Pd表面上[C1、C12上的H原子吸附较为稳定,见图2(a)],吸附后C—H 键的键长增加、键能变弱,表明该脱氢过程始于五元环上靠近N原子的两个C—H键[图2(a) C1、C12处]的断裂。随后8H-NEC、4H-NEC在Pd表面上进一步脱氢至NEC。另外,12H-NEC 的吸附能与Pd 表面结构相关。12H-NEC 在Pd{110}表面的吸附能为-83.2kJ/mol,吸附强度弱于在Pd{111}表面的吸附。这是由于12H-NEC中五元环C1、C12[图2(a)]上的H原子优先吸附在Pd{110}表层Pd原子上,但Pd{110}面上原子暴露密度比Pd{111}面小,表面Pd原子与12H-NEC的五元环中剩余的H[图2(a)中C6、C7]不存在相互作用,因此剩余H 原子需吸附在Pd{110}第二层的Pd原子上[图6(d)],此时形成8H-NEC就需要额外的解吸和再吸附过程(在Pd{111}面上就不需要此过程),因此12H-NEC在Pd{111}面具有优先吸附性。
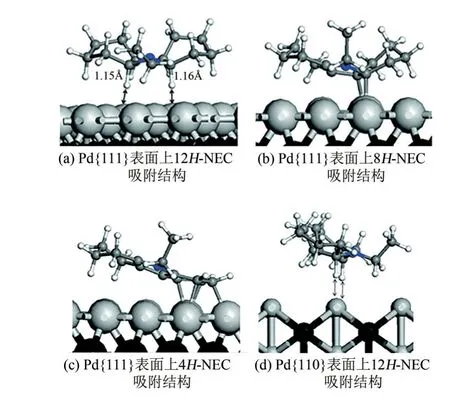
图6 Pd{111}和Pd{110}表面上12H-NEC、8H-NEC和4H-NEC的吸附结构[73](1Å=0.1nm)
2 含氮LOHCs加氢/脱氢催化剂
在相同反应条件下,含氮LOHCs体系的加氢、脱氢反应速率与所采用的催化剂密切相关[11]。含氮LOHCs 加氢、脱氢反应采用的催化剂主要是负载型金属催化剂,反应活性中心包括贵金属(Ru、Rh、Pd、Pt等)和非贵金属(主要是Ni)。催化剂载体主要起分散活性金属的作用,包括Al2O3[25]、活性炭[74]、SiO2[25,75]、TiO2[66,76-77]、还原氧化石墨烯(rGO)[78-79]、水 滑 石[80]、碳 纳 米 管(CNTs)[81]、SAB-15[40]等。另外也可使用负载型双金属催化剂以改善催化性能或利用金属之间的协同作用减少贵金属的负载量[42,82-83]。催化反应过程涉及的加氢反应器主要为釜式反应器和固定床反应器[35,84],脱氢反应器包括喷雾脉冲反应器、固定床反应器、搅拌釜式反应器、管式反应器和变压反应器等[23,85]。
2.1 贫氢分子加氢催化剂
2.1.1 活性位的影响
对于含氮LOHCs 体系来说,贫氢分子的加氢反应通常使用负载型Ru 或Rh、Pd、Ni 催化剂在120~180℃、5~7MPa 的条件下进行[25,41-43,66,86-88]。贫氢分子加氢催化剂主要为负载型贵金属催化剂,如Ru、Rh、Pd等,而使用最多的是Ru[22,46,58,78]。此外,双金属加氢催化剂也有优异的催化性能[2,43,83]。
为探究不同活性位对加氢反应的影响规律,Eblagon 等[60]采用不同金属催化剂对NEC 进行加氢反应,催化活性表现出Ru>Pd>Pt的规律,与金属d带中心和Femi 能级的相对位置[1.41(Ru)<1.83(Pd)<2.25(Pt)]呈负相关关系,如图7所示。这是由于金属d带中心离Femi能级越近,越有利于底物化学吸附及进一步的加氢反应[89]。
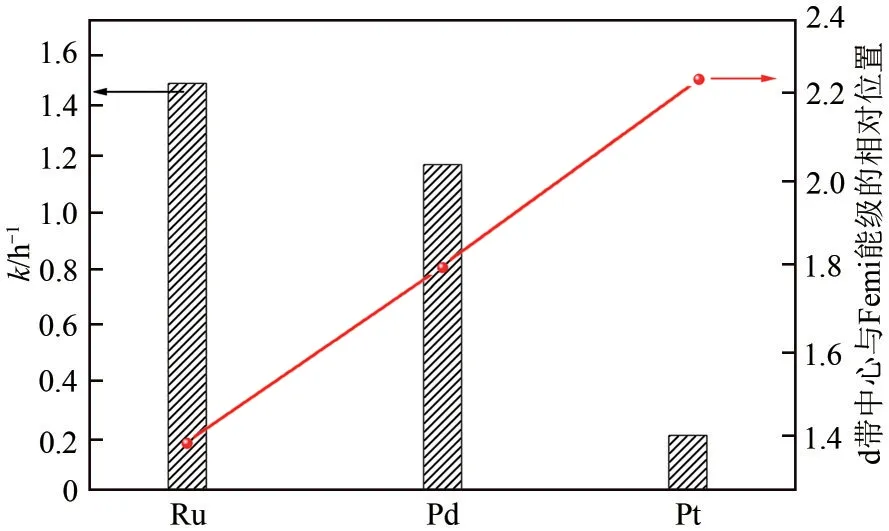
图7 不同金属上NEC加氢反应速率常数对比[60]
将Ru 负载于载体上可以减小金属粒径,从而提升催化剂活性。Shi 等[80]采用Ar 辉光放电等离子体法在水滑石上负载5% Ru纳米颗粒(NPs)并用于NEC 的加氢反应(120℃、6MPa、1h),Ru NPs在该载体上的平均颗粒尺寸(1.86nm)小于其他类型载体上的颗粒尺寸(2.52~3.37nm),因此催化性能最优,在1h内NEC转化率和12H-NEC选择性分别高达100%和99.06%。此外Ru 在载体上金属颗粒的大小还与催化剂的制备条件(超声功率和时间) 有 关,如Liu 等[90]在NiFe 水 滑 石 载 体 上(NiFe-LDH)负载Ru 纳米颗粒(5%Ru NPs/NiFe-LDH),在最优制备条件(超声功率300W、超声时间1h)下Ru NPs平均粒径为1.6nm(在其他制备条件下Ru NPs 颗粒尺寸分别为1.90nm 和2.34nm),表现出优异的催化NEC 加氢性能(110℃、6MPa、80min, NEC 转 化 率100%, 12H-NEC 选 择 性98.88%)。
贫氢分子加氢催化剂的活性中心除贵金属外,非贵金属如Ni 基催化剂也得到研究[25]。常用的Ni催化剂包括负载型Ni催化剂、Raney-Ni 等[59,91]。相比贵金属催化剂,Ni 基催化剂以其低成本在工业应用中备受青睐[25,92]。但是Ni基催化剂与贵金属相比,催化活性较低,并且生成的副产物较多[11]。例如,负载5% Ni 的催化剂对NEC 没有催化加氢活性,需要使用高Ni 负载量的催化剂[56]。研究发现,70% Ni/Al2O3-SiO2催化剂能够使NPC和NEC分别在45min、1.5h 内完全加氢(150℃、7MPa),其活性与贵金属催化剂相当[25]。
2.1.2 载体的影响
活性位负载于载体上往往会与载体形成相互作用,界面处电荷的转移会影响活性位电子结构,从而影响表面吸附和反应过程。因此,载体特性会影响贫氢分子的催化加氢过程。Eblagon 等[56]将Ru 负载在不同载体上(SiO2-Al2O3、沸石、石墨)并用于NEC 的加氢反应(130℃、7MPa),结果表明在活性中心颗粒尺寸和金属负载量相同的条件下,最终产物收率随着载体酸量的降低而降低(酸性载体可接受并转移H 自由基以促进H 溢流[93])。另外,Ding 等[25]将Ni 负载在Al2O3、Al2O3-SiO2、SiO2载体上制备出一系列催化剂并用于NPC、NEC 的加氢反应。结果表明,不同载体的催 化 活 性 顺 序 遵 循AlSiO-1/1>AlSiO-1/3>SiO2>Al2O3>AlSiO-3/1(X/Y表示Al/Si 摩尔比),这是由于复合载体中SiO2可以降低尖晶石NiAlO4的形成并有利于Ni 还原,而Al2O3可以阻止Ni 的团聚并提高金属分散性。
载体的晶体结构类型也会显著影响加氢性能。Yu 等[66]制备了Ru/TiO2催化剂并用于NEC 加氢反应,发现当使用P25(金红石/锐钛矿相约为4/1)作为载体时,催化剂的加氢活性要优于单独使用金红石相或锐钛矿相作为载体的催化剂。Ru 与混合相载体P25 相互作用使得Ru 的颗粒尺寸更小,从而提高了加氢活性和12H-NEC的收率。
2.1.3 双金属协同效应
在负载型催化剂中引入第二金属是提高其活性、降低贵金属催化剂用量的重要方法[75]。在负载型加氢催化剂中,通过引入第二种金属可以改变原有金属的电子结构和空间排列,从而调控催化剂活性[94]。Liu等[43]研究发现引入金属Rh后可降低Ni颗粒尺寸。当使用单金属Ni 催化剂时,反应进行到8H-NEC后,由于平台位点的吸附不再有利,此时8H-NEC 会从活性位点上脱附;而引入第二金属Rh 后,Ni 颗粒尺寸减小并形成更多的Rh/Ni 边缘和台阶位点,这有利于中间产物8H-NEC的吸附及进一步加氢。所制备的0.1% Rh负载量的Rh-Ni催化剂可达到1% Rh 催化剂的加氢性能。另外,Yu等[66]发现双金属Ru-Ni/TiO2催化剂对NEC 加氢时,在Ru、Ni 两种金属表面会发生H 溢流,这样被吸附的NEC 分子可以连续加氢生成12H-NEC 而不受“解吸-再吸附”的影响,从而提升催化活性和全氢产物选择性。
除了构建双金属合金结构以外,也可以通过修饰第二金属物种来提升催化剂性能。Yang等[86]采用机械混合方法用YH3对Pd/Al2O3进行表面修饰,YH3修饰结构在加氢过程中发挥了促进氢气分子和有机物之间氢转移的作用,从而使在相同条件下NEC 的加氢速率提高了7 倍。另外,在Ni/Al2O3催化剂上修饰YH3也能显著提高催化活性,在相同条件下能使催化剂的加氢速率提升34倍[95]。
2.1.4 反应条件对催化性能的影响
贫氢分子加氢是放热、分子物质的量减少的反应,降低温度和增大反应压力可以从热力学上使化学平衡正向进行。但从动力学角度出发,增加反应温度可以增大反应速率。例如,在NEC 加氢反应中,增加反应温度和压力均可以增加反应速率,缩短达到反应平衡所需时间[83],但反应温度过高会导致贫氢分子饱和储氢量降低,其原因除了热力学方面的影响之外,温度过高还会导致催化剂失活[91]。因此对于加氢反应来说,需从热力学和动力学方面平衡反应所需温度,一般选取的催化加氢温度范围为120~180℃[25,41-43]。在一定范围内增加反应压力可以增加反应速率,但压力达到一定值之后(6MPa左右)对反应速率已无明显的影响[82]。此外,在一定温度、压力下,催化剂用量[91]、催化剂活性金属的负载量[96]也会影响加氢反应速率。
2.1.5 加氢催化剂稳定性
除了催化剂活性之外,稳定性也是评价催化剂性能的重要指标。文献报道的用于含氮LOHCs 的加氢催化剂大多具有较好的稳定性。例如,Qin等[82]将高活性Ni-Ru合金纳米颗粒负载在石墨化碳上制得Ni0.5Ru4.5/pg-BC催化剂并用于NEC的循环加氢反应(130℃、6MPa),在10 次连续的加氢反应后,NEC 转化率仍可达到100%,储氢性能为97.91%。此外,其他对NEC 或NPC 加氢催化剂稳定性的测试也表明,在经过数次加氢循环之后催化剂的储氢活性仍可达到原始活性的97%[25,42,82-83,86,90,97]。
为了进一步确认催化剂在使用过程中是否有活性组分的浸出,在反应一定时间后将催化剂滤除,并在没有催化剂的条件下继续反应。结果表明滤除催化剂后反应不再进行,这证明反应过程中不存在催化剂活性组分的损失[83]。另外,含氮LOHCs中氮原子上乙基的空间位阻作用限制了N与活性位的相互作用(N 原子朝着远离活性位点的方向),能有效避免催化剂中毒[57]。
2.2 富氢分子脱氢催化剂
2.2.1 活性位的影响
含氮LOHCs 富氢分子脱氢催化剂主要是贵金属基催化剂,如Pd、Pt、Ru和Rh等,而非贵金属催化剂(Ni/Al2O3、Cu/Al2O3等)几乎没有催化脱氢活性[98-99]。
不同贵金属催化剂的脱氢活性具有较大的差异。Yang等[53]对比了4种不同贵金属催化剂(5%负载量,质量分数)的12H-NEC 脱氢性能(180℃、0.1MPa),结果表明脱氢催化活性遵循Pd>Pt>Ru>Rh的顺序,反应1h后脱氢量(质量分数)分别为3.3%、2.63%、1.91%、0.68%。根据Sabatier 原理,反应(中间)物和活性位之间相互作用应适中,既不宜过强(活性位中毒)也不宜过弱(反应物活化困难)[100]。相比于其他贵金属,Pd与反应物之间有着适宜的相互作用,因此催化活性最优。另外,反应5h 后4H-NEC 在Rh 催化剂上有大量积累(约90%)并造成NEC选择性降低,但该过程使产物中4H-NEC的浓度增加,从而降低体系熔点,使整个LOHCs 体系在室温下为液相。另外,Wang 等[101]在还原氧化石墨烯(rGO)载体上负载Pd、Pt、Rh、Ru、Au 并用于12H-NEC 的脱氢反应(180℃),发现催化活性Pd>Pt>Rh>Ru>Au,与前述结果类似。
贵金属颗粒的粒径对催化脱氢性能有显著影响。Li 等[102]发现在12H-NEC 脱氢过程中,Pd 的粒径从3.7nm 增加到4.9nm 时,脱氢速率下降35%,表明脱氢反应对粒径大小十分敏感。因此,制备超细和高分散的Pd NPs 是提高12H-NEC 脱氢活性的重要方法[4,40]。此外,适当增加Pd价态(如借助Pd和尖晶石氧表面之间的强电子相互作用形成部分带正电Pd[102])可以降低脱氢产物的吸附强度,同时抑制电子从活性原子到不饱和碳原子的转移,从而提高脱氢活性和稳定性[103]。
贵金属脱氢催化剂的活性位点(如Pd)不仅需要吸附反应物以使C—H断键,还需要吸附H原子以形成H2分子进行解吸[94,104]。这在一定程度上阻碍了反应物的连续吸附,导致脱氢速度较慢。但Fei 等[105]发现,对于Pd/MoO3负载型脱氢催化剂,活性位和载体可发挥协同脱氢作用,从而解决上述反应物和H 的竞争吸附问题,如图8,Pd首先促使C—H 键裂解,而H-随后溢流至MoO3表面并将电荷转移给MoO3形成氢质子(H+),当MoO3上氢质子饱和时,反应形成H2分子并从MoO3表面脱附。

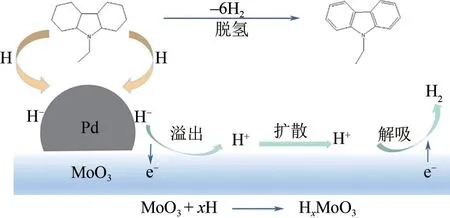
图8 12H-NEC脱氢过程中Pd与MoO3的协同作用机理[105]
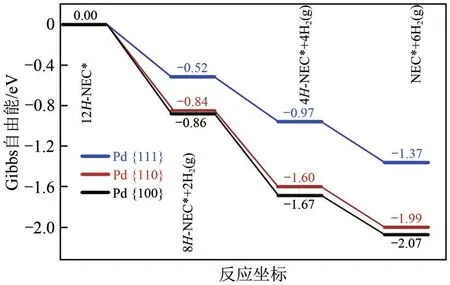
图9 12H-NEC在180℃下Pd{100}、{110}和{111}晶面上脱氢反应的Gibbs自由能[78]
2.2.2 载体的影响
除了活性中心,载体对含氮LOHCs 富氢分子脱氢性能也有重要影响。以Pd 为活性物种,使用不同载体催化剂的12H-NEC 脱氢比活性遵循C>Al2O3>TiO2>SiO2,如图10 所示。C 相比其他载体具有最大的比表面积和孔容,这是Pd/C 催化性能最优的原因。对其他比表面积和孔容相差不大的载体来说,Pd 粒径越小、载体上Pd 的还原度越高,则催化剂的催化性能越好[22]。
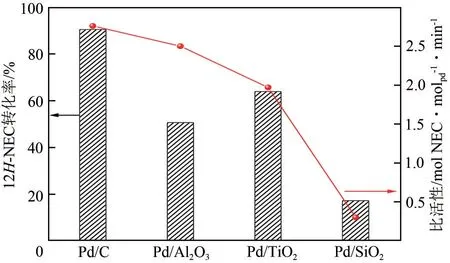
图10 不同催化剂在180℃、1h下12H-NEC脱氢性能[22]
载体与活性中心之间的相互作用也会调控催化剂粒度及电子结构,从而影响催化脱氢性能。如Li等[102]将Pd 负载在4 种尖晶石(MgAl2O4、NiAl2O4、CoAl2O4和ZnAl2O4)载体上并进行脱氢研究。Pd与MgAl2O4之间的强相互作用使Pd颗粒更小,同时使Pd 的化学价态高于其他催化剂,这既提高了反应活化能力,也降低了脱氢产物的吸附强度,从而更有利于脱氢反应。
另外,载体还会影响不同贵金属的活性顺序。Jiang 等[76]以TiO2为载体制备了Pd、Pt 催化剂(5%负载量) 并用于12H-NEC 脱氢反应(180℃、0.1MPa),发现Pt/TiO2催化的脱氢活性和NEC选择性明显优于Pd/TiO2,这与2.2.1 节所述活性趋势相反[53,71,101,110]。这种差异归因于载体的作用,在脱氢反应过程中Pt/TiO2催化剂中Pt 的XPS 谱峰向低结合能的方向偏移,但Al2O3载体上的Pt 不会发生此类现象,半导体载体TiO2可能为Pt 提供了额外的电荷(与Pt0相比,TiO2上的Pt 变成富电子状态)以促进其脱氢性能[111-112]。
2.2.3 双金属协同效应
与加氢催化剂类似,通过构建双金属位结构也能提升富氢分子催化脱氢性能。对于Pd基脱氢催化剂,引入的第二金属包括Ni、Cu、Co等[75,99]。Gong等[99]制备了Pd-M(M=Co, Ni, Cu)/Al2O3双金属催化剂(两种金属总负载量为5%)并用于12H-NEC脱氢反应(180℃、0.1MPa),反应8h 后Pd3Co1/Al2O3、Pd1Co1/Al2O3、 Pd3Ni1/Al2O3、 Pd1Ni1/Al2O3、 Pd3Cu1/Al2O3、Pd3Co1/Al2O3(下角标表示两种金属摩尔比,总负载量为5%,质量分数)脱氢量分别为90.85%(质量分数,下同)、95.34%、86.7%、84.28%、84.8%、84.8%,均表现出比5%Pd/Al2O3(81%)更好的催化活性,这与活性位适宜的电子结构密切相关。一般来说,活性金属的d带中心越接近Femi能级,催化活性越高[113]。引入第二金属(如过渡金属Ni)后,XPS中Pd 3d电子结合能的增加导致Pd的d带中心向Femi能级移动[99,114],因此脱氢活性会进一步提升[115]。但是,第二金属(如Ni、Cu)含量不能过量,否则将导致脱氢率显著降低[75]。
2.2.4 反应条件对催化性能的影响
富氢分子脱氢是吸热反应,因此提高反应温度有利于平衡的正向移动。同时,升高反应温度可以加快脱氢速率并缩短达到平衡所需时间[40]。但是升高温度不仅会增加能耗,而且温度过高会导致产物裂解,发生严重的副反应。因此,含氮富氢分子脱氢反应的温度一般在160~200℃之间[5,40,77]。
脱氢反应为分子数增加的过程,因此降低反应压力可以提高12H-NEC 的脱氢速率。使用商用5% Pd/Al2O3催化剂在0.1MPa、0.05MPa、0.025MPa下反应6h 后(200℃)的脱氢程度分别为87.2%、92.5%、95.5%108。实际上,常压条件不仅操作简单,而且能获得很好的脱氢性能,因此成为该研究的首选条件[40,102]。
2.2.5 脱氢催化剂稳定性
由于含氮LOHCs 储氢技术需要循环加氢、脱氢,因此开发具有高活性和稳定性的催化剂是实现LOHCs 多次储放氢的关键。但是脱氢催化剂的稳定性要差于加氢催化剂[5,81-83,97,115]。在含氮LOHCs 储氢技术应用中,脱氢催化剂的失活是该技术面临的关键问题之一[116],而催化剂失活原因主要是表面积炭或活性位流失。例如,Wu 等[5]对12H-NEC 连续脱氢6次后发现催化剂的重复使用造成Pd NPs的聚集,活性点的数量减少,因此脱氢效率有所下降(93%)。同时,Feng等[40]发现多次循环后多环芳烃吸附在活性位点上形成积炭并覆盖了活性位点,同样导致脱氢效率下降(94.6%)。近年来,一些含氮富氢分子(主要针对12H-NEC)脱氢催化剂的循环稳定性如表2所示。相较加氢催化剂而言,脱氢催化剂的稳定性会差一些。针对脱氢催化剂失活后的再生问题,研究表明Pd 基催化剂可以在温和的条件下氧化再生(低于600K),在氧化-再活化过程中使用更高的温度很难进一步降低碳含量,因此不利于催化剂再生[117]。
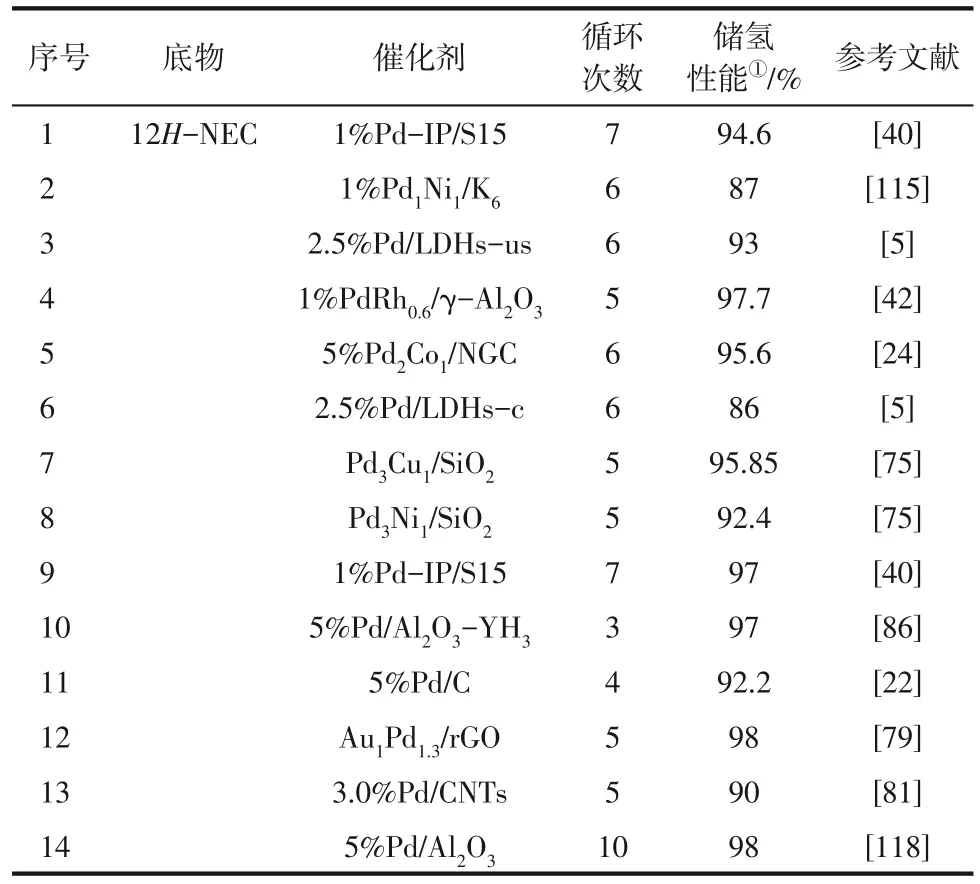
表2 含氮有机储氢化合物脱氢催化剂重复性
3 含氮LOHCs加氢/脱氢动力学
3.1 贫氢分子加氢反应动力学
含氮LOHCs 贫氢分子加氢动力学的文献报道较多,表3汇总了近年来对贫氢分子加氢动力学所采用的催化剂、反应级数及反应活化能。在含氮贫氢分子中,NEC 的加氢反应动力学研究最多,而其他含氮贫氢分子的动力学研究报道较少。
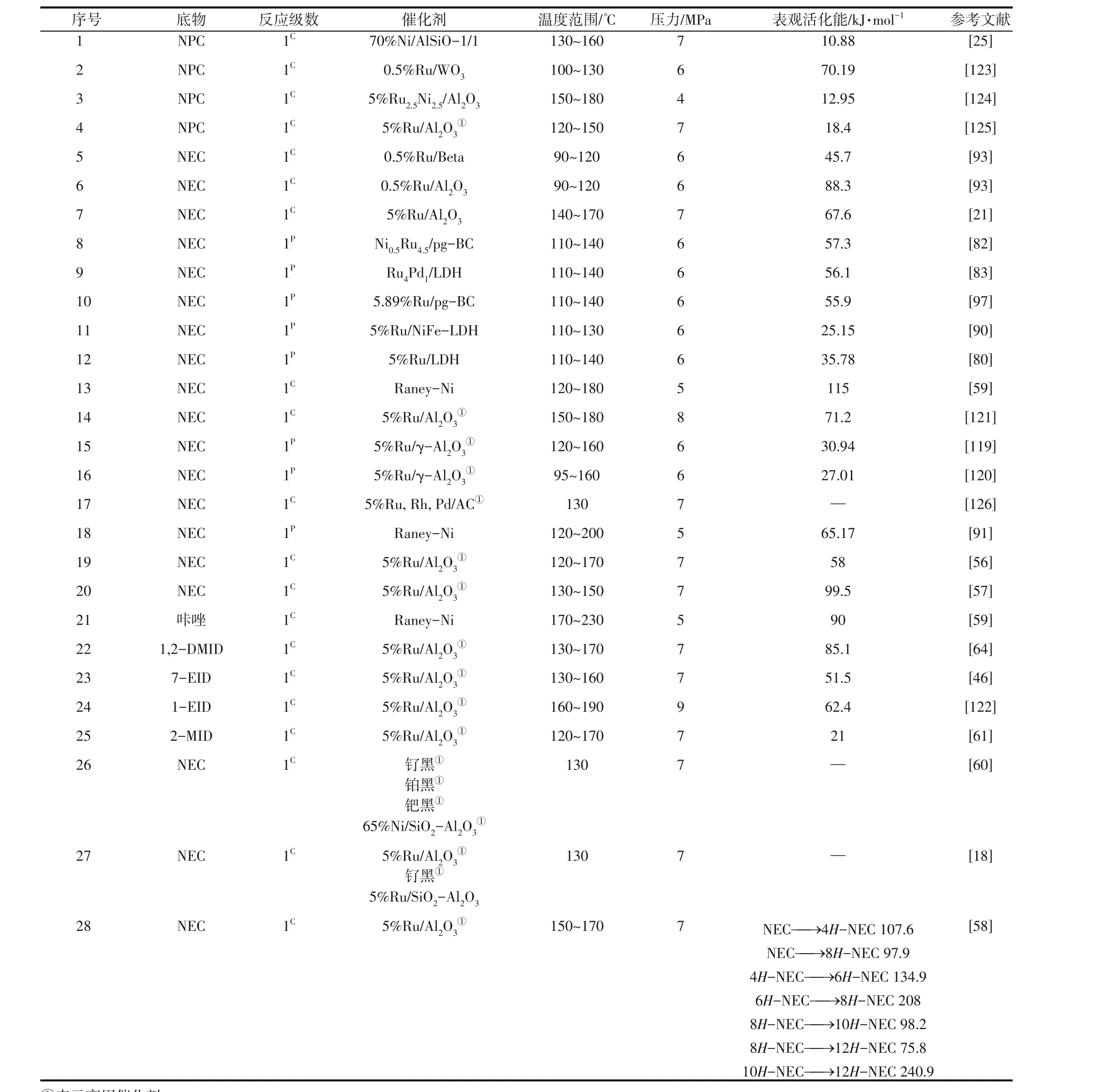
表3 含氮LOHCs贫氢分子加氢反应动力学参数
3.1.1 贫氢分子加氢反应级数
对于含氮贫氢分子的加氢反应级数,目前存在两种观点:一是Sotoodeh 等[57-58]提出的NEC 加氢反应是关于液体反应物浓度的1 级反应(1C),而由于氢气过量(反应通入的氢气量为加氢饱和所需量的2.5倍以上),所以关于氢气浓度为0级反应,即反应速率与反应物浓度成正比,反应物在催化剂表面发生吸附;二是An 等[63,119-120]认为的加氢反应级数是关于液相中氢气浓度的1 级反应(1P),关于底物浓度的0级反应,即反应速率与反应物初始浓度、反应经历的时间无关,而是取决于液相中氢气的浓度,反应物在催化剂表面不发生吸附。表3中贫氢分子加氢采用的多为关于反应物浓度的1级反应,此时认为在加氢反应过程中氢气压力不变而只与反应物浓度有关。
3.1.2 催化连串加氢反应模型

式中,CA0为初始反应物浓度,mol/L;CA、CB、CC、CD分 别 为NEC、4H-NEC、8H-NEC、12H-NEC 的浓度,mol/L;k为反应速率常数,mol/(L·min);t为反应时间,min。

3.1.3 复杂反应网络模型
实际上,含氮贫氢分子加氢反应相比串联路径更为复杂,如图2(a)所示的复杂反应网络包含了平行、连串加氢反应,虽然在计算其动力学时相比连串反应模型更为复杂,但能更精确地获得催化加氢反应动力学参数并区分主次反应路径[58]。
另外,还可以将最终产物12H-NEC 的空间构型也考虑到反应路径中,提出更加复杂的反应网络,如图2(b)所示[60]。若认为各加氢反应步骤均为1 级反应,依据图2(b)的反应网络模型可以得到如式(5)~式(10)所示的动力学方程。
式中,CA、CB、CC、CD分别为NEC、4H-NEC、8H-NEC、12H-NEC 的 浓 度,mol/L;CD_A、CD_B、CD_C为12H-NEC[A]、[B]、[C]构型的浓度,mol/L。
可以看出,相较于连串反应模型,反应网络模型中动力学的计算会增大反应速率常数的求取难度,在解方程式(5)~式(10)时需要不断迭代优化直到满足收敛标准。
3.2 富氢分子脱氢反应动力学
富氢分子催化脱氢的反应物只有液相富氢分子组分,该反应动力学常采用1级连串反应模型。如12H-NEC 脱氢反应动力学,研究采用的是12H-连 串 反 应模型[25,75,99,118]。表4汇总了近年来含氮LOHCs富氢分子的脱氢反应动力学参数。其中大部分研究采用的是简化的表观1级反应模型进行拟合和计算。
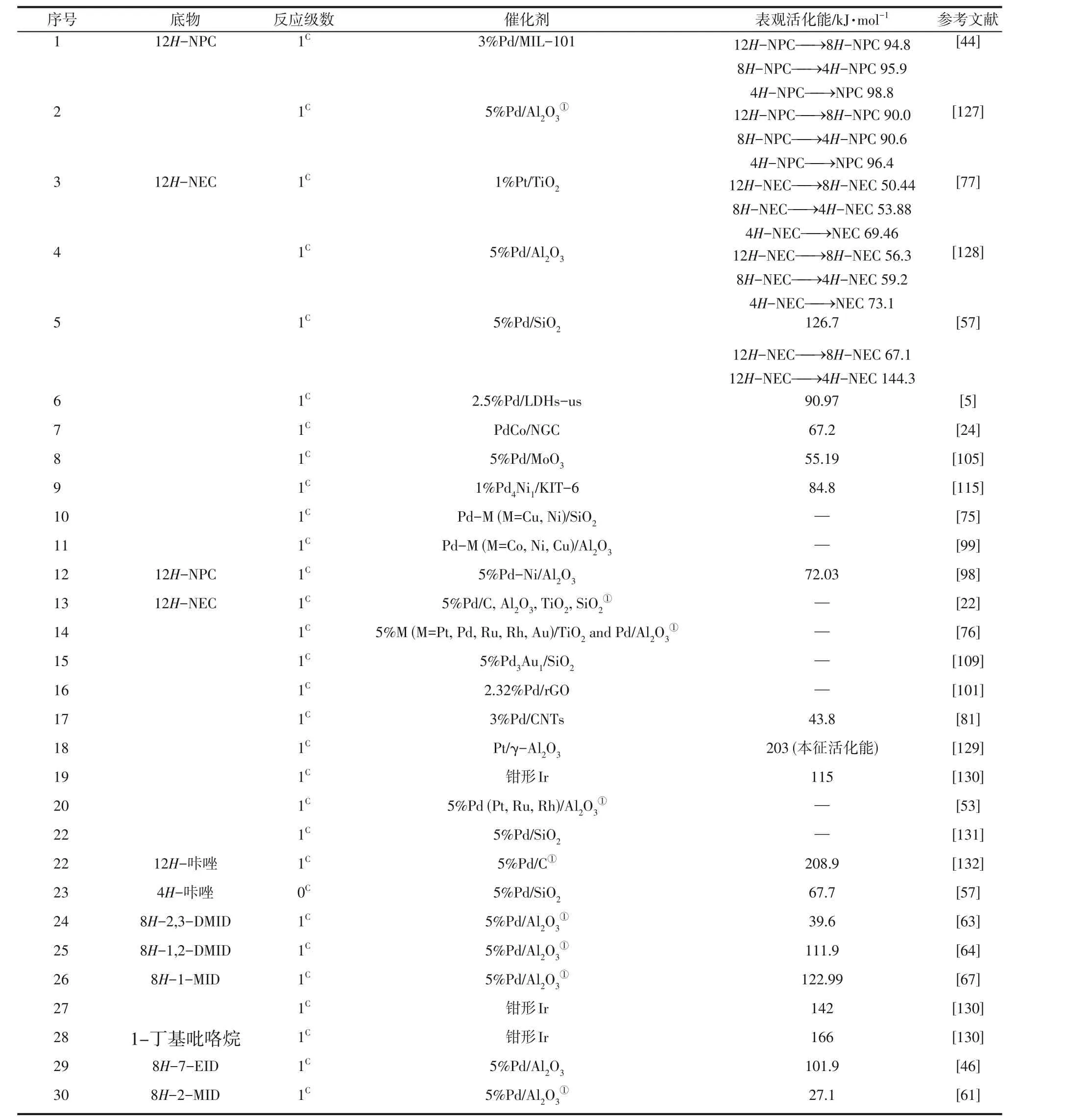
表4 含氮LOHCs富氢分子脱氢反应动力学参数
Sotoodeh等[57]最早对12H-NEC脱氢动力学进行了研究,采用关于反应物浓度的1 级反应计算出12H-NEC 生成8H-NEC 和4H-NEC 的速率常数,(表4 序号5),该过程的动力学方程如式(11)~式(13)所示。
式 中,CD、CC、CB分 别 为12H-NEC、8H-NEC、4H-NEC的浓度,mol/L;k为反应速率常数,mol/(L·min)。由于12H-NEC的扭曲结构,8个C 原子吸附在金属表面所需的驱动力大于4个C原子的吸附,因此12H-NEC 直接脱氢生成4H-NEC 所需能垒更高。
Gong 等[77]使用该模型对1% Pt/TiO2催化12HNEC 脱氢反应动力学进行了研究,得出三步脱氢反应活化能分别为50.44kJ/mol、53.88kJ/mol、69.46kJ/mol,其中,第三步活化能垒最高,为整个反应的速控步骤(表4 序号3)。另外,12H-NPC 脱氢反应路径为12H-NPC→8HNPC→4H-NPC→NPC,其中,4H-NPC 生成NPC为整个过程的速控步骤[44]。全氢-7-乙基吲哚(8H-7-EID)的脱氢过程为8H-7-EID→4H-7-EID→2H-7-EID→7-EID,其中2H-7-EID 到7-EID的转化在动力学上有利,4H-7-EID 生成2H-7-EID 为脱氢过程的速控步骤[46]。对于上述的富氢分子脱氢过程,若采用1级反应,则该过程与3.1.1节贫氢分子加氢连串反应动力学方程式(1)~式(4)一致。此外,全氢-1,2-二甲基吲哚(8H-1,2-DMID)[64]、全氢-1-甲基吲哚(8H-1-MID)[67]的脱氢也是采用逐步脱氢的连串反应模型,但在相关研究中主要采用的是简化的表观1级反应模型。
若采用表观1 级反应模型,12H-NEC 的脱氢能垒范围为43.8~203kJ/mol,全氢吲哚及其同系物的脱氢能垒在39.6~122.99kJ/mol 之间。而采用1 级连串反应模型,12H-NPC 脱氢各步反应能垒分 别 为90~94.8kJ/mol、90.6~95.9kJ/mol、96.4~98.8kJ/mol;12H-NEC 脱氢各步反应能垒分别为50.44~56.3kJ/mol、 53.88~59.2kJ/mol、 69.46~73.1kJ/mol。可见,采用1 级连串反应模型所得到的各步活化能相差范围更小,结果更为准确。
4 总结与展望
本文全面总结了含氮LOHCs体系的研究进展,包括加氢/脱氢过程的反应路径及催化机理、催化剂结构及反应条件对反应的影响规律,基于连串反应、反应网络等模型的动力学以及该体系的相关应用等。含氮LOHCs 贫氢分子加氢催化剂主要为负载型Ru、Ni等催化剂;脱氢催化剂以负载型Pd催化剂为主,但是其稳定性仍有待提升。含氮LOHCs 的加氢、脱氢反应动力学的研究主要采用简化的表观反应动力学模型和连串反应模型,获得了相应的动力学参数。结合理论计算的加氢、脱氢反应机理,揭示了催化反应过程并确定了速控步骤,为催化剂设计、反应工艺优化提供了关键支撑。目前,该体系正在开展放大示范研究和验证,表现出较好的应用前景。然而,该储放氢体系仍面临诸多挑战。
(1)含氮LOHCs 配方优化 如前所述,此类LOHCs 贫氢分子或富氢分子具有熔点高的问题,可采用多种分子复配策略来解决。但是,配方优化对液体理化性质及储放氢性能的影响规律不明确,需要进行深入且细致的研究。
(2)储放氢连续反应体系 目前文献报道的含氮LOHCs 储放氢研究主要集中于间歇反应体系,与连续应用场景仍不能很好衔接。因此,应重点关注成型催化剂在固定床连续反应器上的催化体系研究,为高活性和高稳定性催化剂筛选及工艺条件优化提供关键支撑。
(3)成型催化剂设计与制备 围绕上述连续反应体系,设计并制备低成本、高效的成型催化剂是最为关键的环节之一,特别是关注高活性高稳定性脱氢催化剂的研制。
(4)催化剂结构与产物立体构型及其理化性质的构效关系 加氢或脱氢产物的立体构型可能会严重影响后续的脱氢或加氢反应、化合物的相态(液态或固态)等,同时其立体构型又与催化剂活性结构密切相关。因此,需要解析活性位结构对产物分子立体构型、理化性质甚至后续加氢/脱氢反应性能的影响规律。
(5)更为准确的反应动力学 对于间歇反应,可进一步深入研究加氢/脱氢反应的本征动力学;另外,应开展基于连续反应器的反应动力学研究(同时需要考虑内/外扩散的影响)。
(6)含氮LOHCs 物性数据库构建 该数据库的构建是推动其应用的重要支撑,除了包括基础理化性质外,还要测试毒性、爆炸极限、闪点等性质。
猜你喜欢
中国特种设备安全(2022年4期)2022-07-08
中国特种设备安全(2022年4期)2022-07-08
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13
昆明医科大学学报(2020年12期)2021-01-26
山东化工(2019年11期)2019-06-26
国际呼吸杂志(2019年1期)2019-03-08
现代食品(2016年24期)2016-04-28
信息记录材料(2016年4期)2016-03-11
材料科学与工程学报(2016年5期)2016-02-27
中国资源综合利用(2016年12期)2016-01-22
