
电沉积法制备碱性电解水镍基析氧电极的研究进展
2024-01-16 11:29张静贺业亨王晶晶夏博文赵秦峰王延飞余颖龙邵晨熠龙川
化工进展 2023年12期
张静,贺业亨,王晶晶,夏博文,赵秦峰,王延飞,余颖龙,邵晨熠,龙川
(1 中国石油石油化工研究院,北京 102206;2 中国石油大学(北京)化学工程与环境学院,北京 102249)
氢是一种理想的能量载体,具有质量能量密度高、使用过程环境友好且无碳排放等优点,被认为是21 世纪的终极能源[1-7]。在“碳达峰、碳中和”背景下,氢能作为深度脱碳以及实现清洁能源转型的重要途径,产业已进入高速发展阶段。据中国氢能联盟估算,到2060 年,我国氢气的年需求将超过1.3亿吨,在终端能源消费中占比约20%[8]。各国已发布的氢能战略中,远期(2050 年)均以“绿氢”为主,“绿氢”已成为能源转型与碳中和的重要抓手[9]。利用太阳能、风能等可再生能源电解水制氢,是大规模制取“绿氢”的理想路线。因此,高效率、低成本的电解水制氢技术是未来氢能工业重要的发展方向之一。
电解水制氢技术主要包括碱性电解水制氢技术、质子交换膜电解水制氢技术、高温固体氧化物电解水制氢技术和阴离子交换膜电解水制氢技术等。其中,碱性电解水制氢技术商业化程度最高,已在多个绿氢示范项目中应用,但仍存在工作电流密度低、能耗水平高、氢气成本高等问题。电解制氢系统的用电成本占氢气价格的40%~60%,甚至可达80%。该成本与电解槽槽压直接相关,槽压包括水的理论分解电压(1.23V)、槽部件损失电压、膜损失电压、电解质损失电压、析氢反应(HER)和析氧反应(OER)过电位等[10]。HER 和OER 为电解水过程中的两个半反应,HER 是双电子转移过程,反应动力学快速;而OER 是四质子/电子-耦合反应过程,其动力学过程缓慢且伴随较高过电位[11-13]。因此,析氧电极材料性能的优劣很大程度上影响着电解槽槽压及单位制氢成本。目前商业电解槽广泛使用的析氧电极为纯镍网或泡沫镍,析氧性能有很大提升空间[14]。虽然纯镍网或泡沫镍具有成本低廉、导电率高、强度高、耐腐蚀性能好等优点,但其活性位点的数量及本征活性很难进一步提高。在碱性条件下,镍基催化功能层主要以氧化物/氢氧化物的形式存在,具有较强的OH-吸附能力,H2O分子可以在电极上有效地解离为OH-中间产物,OER 性能优异且稳定性好。因此,镍基材料作为优良的OER 催化剂被广泛研究。在镍网或泡沫镍上复合镍基催化剂是提升镍基自支撑电极OER 性能的有效方式,优化镍基催化功能层及其复合方式是重要的方向之一。
在镍基底(镍网或泡沫镍)上复合镍基催化功能层的方式有很多,包括水/溶剂热法、化学气相沉积法、微乳液法、喷涂法、电沉积法等[15-20]。其中,采用电沉积法制备自支撑电极,即在导电基底上直接生长催化功能层,优化表面电极结构,具有工艺简单、条件温和、利于放大生产等优势,成为工业化生产镍基OER电极的理想工艺之一。
本文首先简要介绍了电解水析氧原理和电沉积方法,然后从过渡金属镍的(氢)氧化物、非贵金属元素的掺杂以及非金属元素的掺杂3个方面总结分析了近年来采用电沉积技术制备用于碱性电解水镍基析氧电极的研究成果(图1为镍基自支撑电极OER性能的影响因素),采用不同电沉积工艺可以调控影响镍基自支撑电极OER 性能的关键因素。通过增强催化剂的电导率及金属间的协同作用、增加活性位点数量、减小扩散路径以及改变表面原子构型来增强OER 性能,最后为后续研究的工作重点做出总结并为未来工业化提出建议和展望。
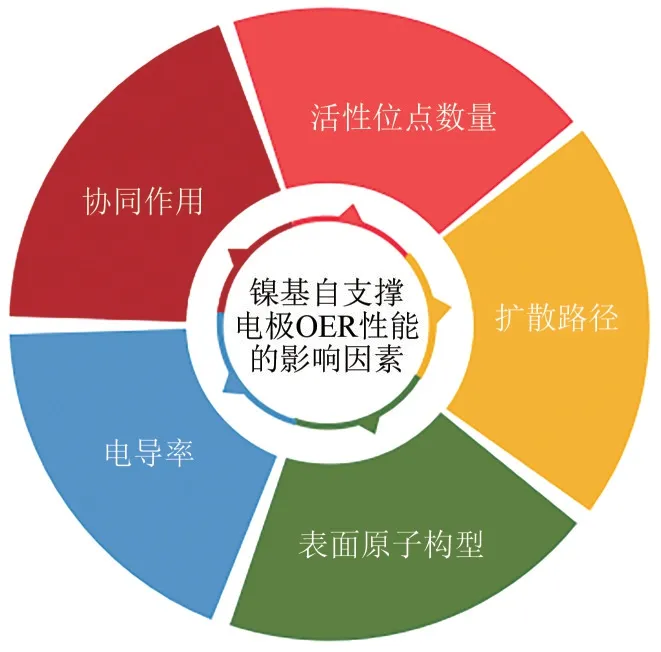
图1 镍基自支撑电极OER性能的影响因素
1 电解水析氧原理和电沉积法
1.1 电解水析氧原理
在碱性条件下,电解水反应示意图如图2 所示,阴极发生析氢反应,阳极发生析氧反应[21]。电解过程中,阳极上OH-失去电子生成H2O和O2,化学反应如式(1)所示。
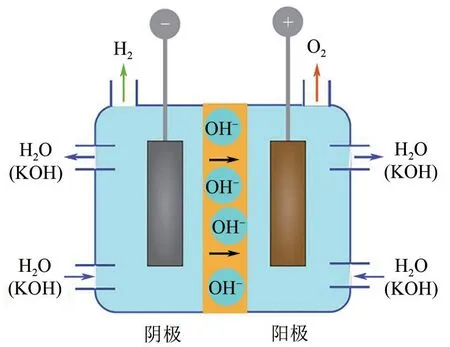
图2 碱性电解水反应示意图[21]
从微观上来讲,OER 反应包括电极表面反应物种的吸附、电子的转移以及反应物种的脱附过程。OER 反应涉及多电子转移过程,目前主要存在两种反应机制:吸附演化机理(AEM)和晶格氧氧化机理(LOM)[图3(a)和(b)]。根据AEM,OH-首先吸附在表面O空位上,被吸附的HO*随后发生脱质子反应形成O*,接下来的O—O键形成步骤中O*与另一个OH-反应形成HOO*中间体,最后通过HOO*的脱质子化和活性位点的再生生成O2;与AEM不同的是,LOM在两个相邻的金属位点上进行反应,金属位点上的两个HO*发生脱质子反应,生成两个金属氧基(M-O*),接着这两个相邻的O*直接结合形成O—O 键,而不是与水或OH-结合形成HOO*,最后O2生成,2 个空的金属中心被OH-占据[22]。
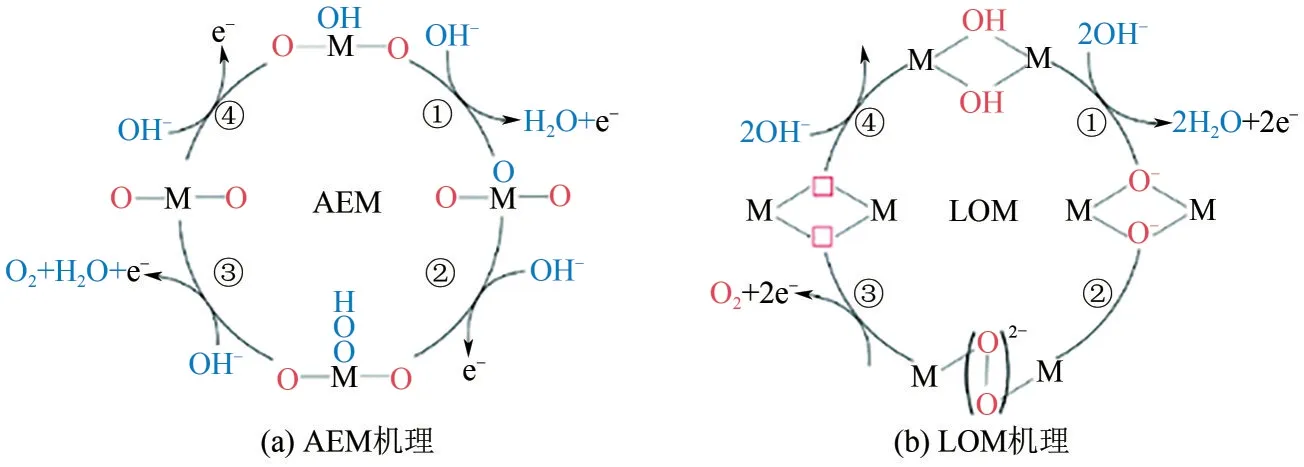
图3 OER反应机理[22]
从动力学方面来说,电解水OER 反应主要是由电催化剂与OER 氧中间体(HO*、O*、HOO*)之间相互作用驱动的,电催化剂与氧中间体之间的吸附结合能可以作为评价催化材料OER 活性的标准。根据Sabatier 原理[23],当电催化剂表面与O 的结合作用太弱时,不易形成中间产物HO*;当电催化剂表面与O的结合作用太强时,不利于HO*进一步反应生成HOO*;电催化剂与氧中间体之间适度的结合能,才有利于中间体的吸附和解吸,从而获得较高的OER 催化活性。不同电催化剂的结合能和活性(通常用过电位表示)之间的关系可以简化为OER反应火山图,如图4所示,顶点代表最佳OER 催化活性。可以看出,非贵金属Ni 的氧化物处于靠近顶点的位置,是高性能OER 催化剂中理想的组分[24]。
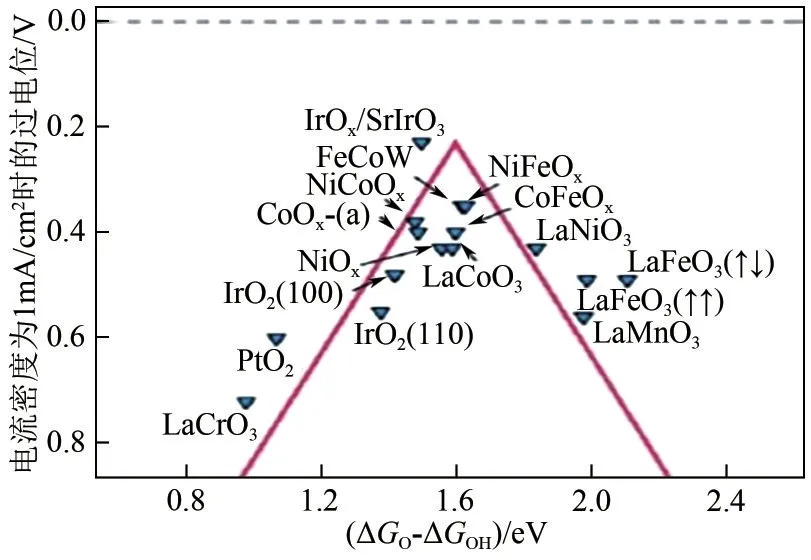
图4 OER反应火山图[24]
1.2 电沉积法
电沉积法是基于电镀中的金属离子沉积技术,在外部电场的作用下,电流通过电解质溶液中正负离子的迁移并在电极上发生氧化还原反应而形成镀层的技术。在阴极(工作电极)获得电子,产生金属离子的还原,是制备金属颗粒和金属层状结构的有效方法。
1.2.1 金属电沉积原理
金属电沉积的阴极过程一般由液相传质、前置转化、电荷传递和电沉积过程串联组成[25]。在电解质溶液中,电流以离子的形式流动,阴离子被吸引到阳极,金属离子被吸引到阴极还原电沉积下来。阴极过电位是电沉积过程进行的动力,只有阴极极化到金属析出电位时才会发生金属离子的还原反应[25-26]。阴极还原的金属原子聚集形成晶核,进而生长为晶体。只有达到一定的阴极极化值(析出电位)后,晶核才能稳定存在,否则会重新溶解。图5表示金属离子在水溶液中还原的可能性,只有金属元素在周期表中处于恰当位置时,在阴极上还原电沉积的可能性才越大,越靠左边或越靠右边则更难以实现。
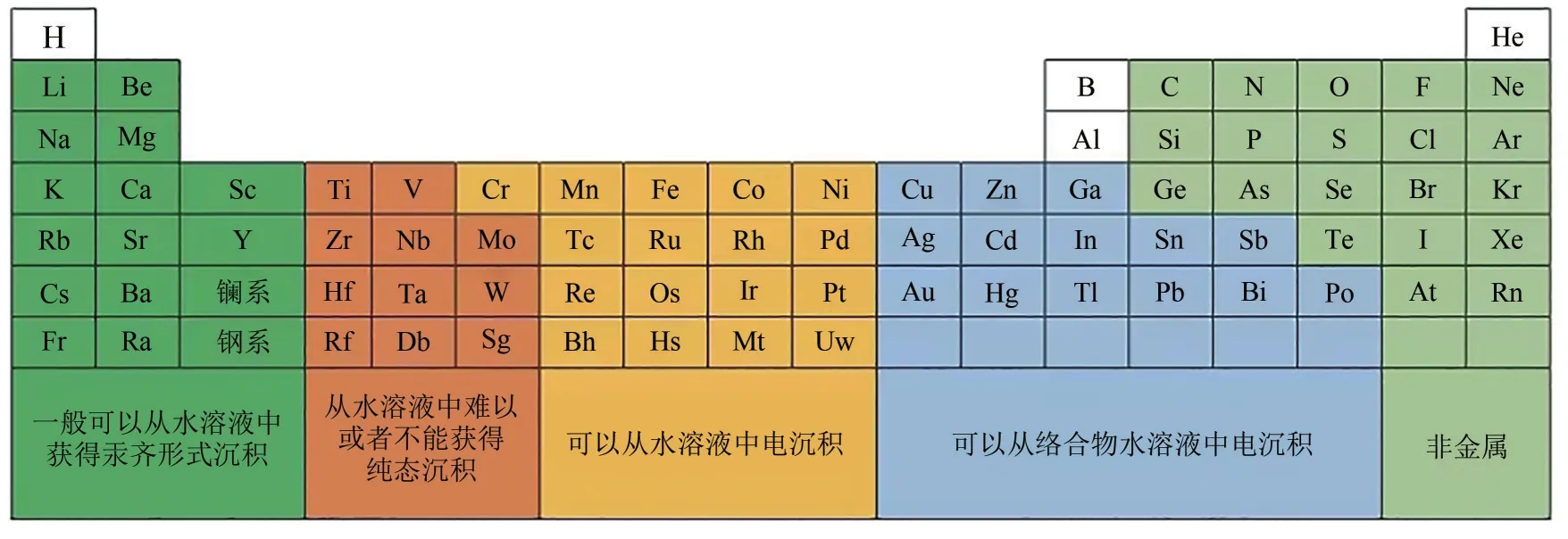
图5 在水溶液中金属离子阴极还原的可能性[25]
1.2.2 电沉积工艺
电沉积工艺中,恒电流电沉积法、恒电位电沉积法、共电沉积法及改进的水热电沉积和微波电沉积法都受到广泛关注[15,27-30]。目前最常用的是恒电位电沉积法和恒电流电沉积法,恒电位沉积通常在三电极体系中进行[图6(b)],恒电流沉积可以在两电极体系中进行[图6(a)][31-32]。实验阶段的电沉积过程,主要使用电化学工作站(或直流电源)和电沉积池。如果沉积过程在较大的电沉积池中进行,通常还需要电解液循环泵、加热恒温及搅拌装置。电沉积流程一般包含基底前处理、电沉积过程以及电沉积后处理。基底前处理通常是将基底上的氧化物、有机物等杂质通过酸和有机溶剂进行处理,沉积过程中通过控制施加的电位或沉积电流,溶液中的反应物被氧化/还原,连续沉积在工作电极上,工艺参数包括电流密度、沉积电位、沉积时间、沉积电解液温度、溶液黏度以及搅拌速度等,可通过调整参数中一项或几项来控制晶体的成核与生长速率,从而获得不同的功能层形态和尺寸。最后将沉积后得到的不同样品进行后处理,如清洗表面杂质、(真空)干燥等步骤,随后得到产物。
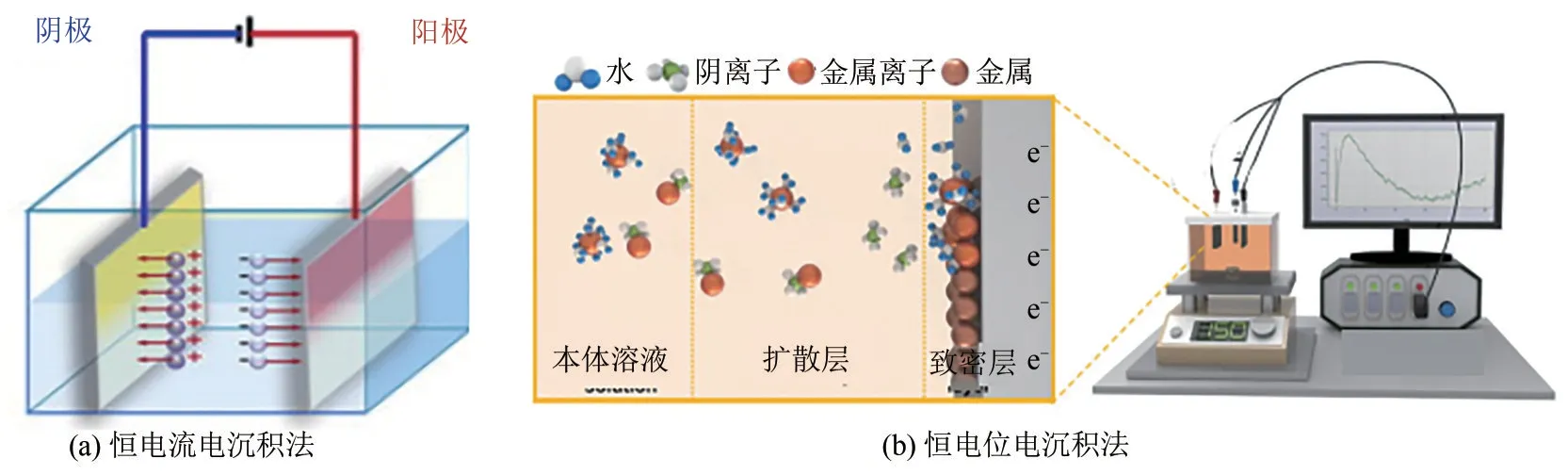
图6 电沉积工艺示意图[31-32]
2 电沉积法制备镍基析氧电极
在镍网或泡沫镍上复合镍基催化功能层的方法有很多。与电沉积法相比,采用其他方法(如水/溶剂热法、化学气相沉积法、微乳液法、喷涂法等)制备镍基自支撑电极的过程中,通常需要使用表面活性剂或封盖剂调控形貌结构,或涉及高温高压反应条件、形貌结构不易控制。采用电沉积法在导电基底上直接生长镍基催化功能层,具有如下优势:工艺快速简便、生产效率高且成本低廉;可以精确控制电极的大小、结构,可以通过改变前体溶液的种类来生产不同类型的催化功能层;并且催化功能层的形貌和组成易于调控,易于获得均匀分散的电催化剂,制得的电极重现性好;不需要额外的封端剂或还原剂,易实现工业规模化制备[33-36]。下文综述了采用电沉积法在镍基基底上制备得到不同类型的催化功能层的研究进展,包括单金属镍(氢)氧化物、双金属及多元金属镍基催化剂和非金属元素掺杂的镍基催化剂,并在表1中对不同类型的镍基自支撑电极的OER性能进行了总结。

表1 文献中报道的电沉积法制备镍基电催化剂的OER性能
2.1 单金属镍(氢)氧化物自支撑电极
镍的氧化物[37-38]和氢氧化物[27,39-40]具有特殊的电子结构、稳定的化学性质和较高的本征活性,是很有前途的OER 催化剂。通常来说,催化剂活性位点的数量和本征活性是影响其催化活性的关键要素。通过优化电催化剂形貌、尺寸能有效暴露更多的表面活性位点、增加配位不饱和原子数目,从而提高催化剂活性。此外,电催化剂孔隙率及表面微观结构参数会影响电解质渗透、气体产物释放过程。
针对催化功能层为氢氧化镍/氧化镍的镍基自支撑电极,已有研究采用电沉积技术来调控氢氧化镍/氧化镍的形貌、尺寸和表面微观结构,以提高电极的比表面积和增加活性位点数量,促进其OER性能。Yao等[40]研究了NiCl2电解质浓度对纳米颗粒的晶型结构和OER 性能的影响。当NiCl2的加入量小于9mmol时,得到立方晶型Ni;当加入量达到9mmol后,菱形晶型的α-Ni(OH)2·0.75H2O/NF开始出现。此外,催化功能层的厚度会随着电沉积时间的延长而增大,当NiCl2的加入量为10mmol,沉积时间依次为10min、20min、40min和60min时,所得催化功能层的厚度分别为1.2mm、2.2mm、3.7mm和5.8mm。其中沉积40min 得到的α-Ni(OH)2·0.75H2O/NF 电极在电流密度为100mA/cm2时,过电位为357mV。Wu等[27]通过恒电流(2mA/cm2)沉积技术在导电性良好的泡沫镍/氮掺杂的碳纳米管(NF/NCNT)混合衬底上合成了α-Ni(OH)2纳米片,在1.0mol/L 氢氧化钾电解液中,电流密度为10mA/cm2时,过电位为254mV。但上述基底上制备的氢氧化镍催化剂为纳米颗粒或较厚的纳米片,OER 活性较差,可以通过改变电沉积条件进一步优化氢氧化镍/氧化镍催化功能层及其OER性能。Cao等[39]利用较高的沉积电位在NF上沉积了二维超薄Ni(OH)2纳米片,交联构成了三维花瓣状团簇,并研究了电沉积条件(电沉积电位、电沉积时间和电解质浓度)对Ni(OH)2纳米片OER 性能的影响。在-2.0V(vs.Ag/AgCl)的沉积电位,0.2mol/L 的电解质浓度下电沉积300s时,得到的α-Ni(OH)2纳米片的厚度仅约为3.0nm,并且形成了三维花瓣状团簇结构,使得电极具有较大的比表面积和高度暴露的催化活性位点,促进了电子传导/质量传输,从而改善了α-Ni(OH)2/NF 电极的催化性能。在10mA/cm2和100mA/cm2的电流密度下,α-Ni(OH)2/NF 电极的过电位分别为192mV 和240mV。此外,α-Ni(OH)2/NF电极甚至可以在低过电位下驱动高电流密度(500~1000mA/cm2),极具工业化应用前景。电沉积时间和电解液浓度对基底上镍基催化功能层的负载量有明显影响,随着沉积时间的增加和电解液浓度的增大,负载量随之增大。适中的负载量才会提升电极的OER 性能:负载过少,活性位点数量较少;而负载量过大,则会影响活性位点的暴露,增加电荷转移阻抗,导致电导率降低、OER 活性较差。
采用电沉积技术控制材料的微观结构,促进纳米材料孔道、比表面积的增长,可大大影响电子、电解液、气体的扩散路径以及活性位点数量的增长。Yu 等[37]采用电沉积技术,在Ni(NO3)2乙腈溶液中将活性Ni纳米点团簇(NiND)沉积在NF上,如图7(a)所示,在NF 基底上电沉积得到的NiNDs 催化剂呈现多孔结构,暴露了更多活性位点,在空气中干燥后,生成NiO/NiND 复合材料,得到无黏结剂和无杂原子的镍基自支撑电极。图7(b)透射电子显微镜图像和高分辨透射电子显微镜图像显示了纳米颗粒堆积形成的多孔结构,孔道尺寸在3~8nm之间,该结构有利于电解质及气体的渗透和扩散,同时进一步证明沉积到NF 上的催化功能层由2nm左右的NiO 和Ni 纳米颗粒组成,可以提供快速的电子转移路径。氮气吸脱附实验结果表明NiO/NiND 复合材料的比表面积为69m2/g,暴露了更多的活性位点。该电极在电流密度为50mA/cm2时,过电位为360mV,且具有较高的催化效率(法拉第效率接近100%)和长期稳定性(13mA/cm2的电流密度下保持20h 而无衰减)。在此基础上,将电极放大制备到80cm2,同样可以在13mA/cm2的电流密度下保持20h而无衰减,这说明放大制备在大规模的电解水中具有很高的工业应用潜力,但大面积电极测试过程中,由于溶液电阻、接触电阻等外电阻过大,使得电解电压增大较多。
2.2 双金属及多元金属镍基自支撑电极
不同的金属组合可以调控镍基催化剂的表面原子构型和反应过程中氧中间体的吸附能、增强材料导电性能、增加活性位点数量、促进金属间的协同作用,因此构建不同金属组成的镍基催化功能层是提高镍基电极电催化性能的有效方法[19,28,41-44]。相比于单一金属的镍基自支撑电极,电沉积法在制备金属元素掺杂的镍基自支撑电极上更具有优势,只需改变沉积电位和沉积电解液类型,就可用简单的设备和较低的生产成本获得在许多条件下无法通过其他制备方法得到的双金属或多金属镍基自支撑电极,制备时间也大大缩短。
在催化功能层为双金属的镍基自支撑电极中,Fe 元素的掺杂在20 世纪就已开始被研究,可以大大提升镍基自支撑电极的OER 性能[45-46]。Li 等[47]制备了NiFe 层状双氢氧化物(LDH)电极,电流密度为10mA/cm2时,过电位为283mV。Zhou等[48]在不改变沉积总电荷量的情况下,分别在250mA/cm2、500mA/cm2、750mA/cm2的强阴极极化电流密度下,采用超快速电沉积法在镍网上直接生长氢氧化镍铁材料,得到的NiFe(OH)x-250/Ni、NiFe(OH)x-500/Ni、NiFe(OH)x-750/Ni电极均具有优于贵金属RuO2催化剂和镍网电极的OER性能[图8(d)],证明Fe元素掺杂后促进了Ni 基电极OER 性能的提升。此外,研究表明,沉积电流密度对NiFe(OH)x/Ni 电极催化功能层的纳米结构有较大影响,如图8(a)~(c)所示,当电流密度为500mA/cm2时,NiFe(OH)x纳米片在Ni 网表面生长形成花状结构,纳米片之间的孔径为100~200nm,有利于离子、水和气泡的传输。除沉积电流密度外,沉积电解液中阴离子种类的改变也会大大影响催化功能层的纳米结构,例如相比于硫酸盐,氯离子可以取代吸附在工作电极表面的氧,防止电极钝化后阻碍Ni 和Fe 的共沉积。Jin等[43]通过一步电沉积法在不同阴离子(Cl-、SO42-)镀液中合成NiFe/NF电极,研究发现在Cl-溶液中沉积得到的NiFe/NF电极比在SO42-溶液中沉积得到的电极具有更高的OER 催化活性。这是由于在SO42-溶液中沉积动力学较慢,沉积得到的颗粒尺寸较大,使得电极具有较小的比表面积和较少的活性位点。而在Cl-溶液中沉积得到的催化功能层,具有丰富的孔隙结构,有利于OER 过程中电解液和气体的扩散。
此外,研究人员相继用电沉积法制备了其他金属元素掺杂的双元镍基自支撑电极。Etesami 等[49]采用超快速电沉积技术,在NF 上合成了NiFe、NiCo、NiCu 等双金属LDH 结构,皆呈短程有序结晶或非晶结构。该LDH 结构不仅可以缩短气体和电解液的扩散路径,而且平衡了整体电荷并增加了层间间距,大大增加了镍基电极的电化学活性面积(ECSA),在电流密度为10mA/cm2时,NiFe、NiCo、NiCu的过电位分别为495mV、524mV、603mV。
近年来,许多研究采用电沉积法制备了多元金属镍基自支撑电极。Huang 等[50]以NF 为基底,在600mA/cm2的恒电流下得到了NiFeMo/NF 自支撑电极,通过电化学性能测试,在电流密度为10mA/cm2时,过电位为306mV,比IrO2和NiFe/NF 的过电位分别减少了71mV和28mV。NiFeMo/NF拥有较优的性能是因为Mo原子的掺杂会导致Ni晶格膨胀,同时促进了电子从Mo向Fe和Ni的转移,三金属之间的强相互作用可以促进OER 反应过程中电子、电解质的传输,加速OER 反应动力学;此外,该电极由于没有聚合物粘结剂,确保了催化功能层和NF 基底的紧密接触,防止生成的气体使催化层脱落;最后,Mo的掺入显著提高了ECSA和活性位点的数量,从而加快催化反应的进行。Wu 等[44]采用恒电流电沉积方法在NF 上制备了自支撑Ni-Fe 和Ni-Fe-Sn 薄膜,Sn 元素的引入使Ni-Fe 薄膜由致密的光滑表面转变成为粗糙的花椰菜状结构,为反应提供了丰富的活性位点;同时,Ni、Fe 和Sn 之间的强相互作用增强了电子传输能力,进一步优化了OH-吸附和O2解吸的能力,从而提高了镍基电极的OER 性能。Wang 等[28]通过恒电位电沉积法精细调节Co 元素在NiCoFe-LDHs 催化剂中的比例,制备了具有独特纳米阵列结构的Ni(Co0.5Fe0.5)/NF 电极。通过与水热法和共沉淀法制备的电极相比,电沉积法制备的Ni(Co0.5Fe0.5)/NF 电极在电流密度为10mA/cm2时,过电位为209mV,具有更加优异的OER 性能。这是由于Ni(Co0.5Fe0.5)/NF 电极有更高的Co3+/Co2+比,暴露了更多的缺陷结构,不仅大大增加了电化学活性位点数量,还调节了中间体的吸附自由能,降低了反应能垒;电化学阻抗测试表明该电极有较小的电荷转移电阻,有利于析氧反应过程中的电荷转移,大大增强了电极的导电性。Bo等[20]通过恒电位电沉积法在NF 衬底上合成了非晶态三金属NiFeCr(氢)氧化物,其OER 性能优于NiFe(氢)氧化物电极。引入Cr元素后,镍基电极性能的提升除了与非晶态结构产生的丰富的不饱和位点有关,还与电极表面Ni、Fe、Cr 位点的原子构型和协同效应密切相关[图9(a)]。通过密度泛函理论计算证实,在NiFeCr 催化剂中,Cr,Fe-subβ-NiOOH 表面中的Cr 原子是活性位点;在过渡金属羟基氧化物中,Ni、Fe、Cr 三种金属位点与氧中 间 体 的 结 合 能 力 为Cr>Fe>Ni,Cr,Fe-subβ-NiOOH 表面的Cr 原子与氧中间体之间具有最佳的结合强度[图9(b)],Cr被氧化为Cr6+,该位点的析氧过电位低于NiFe羟基氧化物中Fe3+位点的析氧过电位,从而促进了镍基电极OER性能的提升。
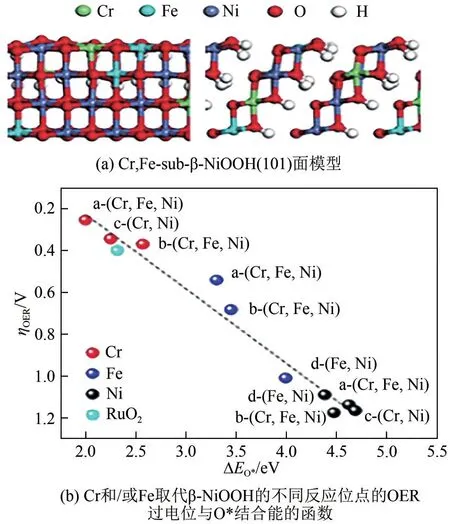
图9 NiFeGr/NF的原子构型[20]
相比于晶体结构材料的高导电性和可控的活性面积,低结晶度或非晶态材料具有丰富的不饱和配位位点,在电催化过程中可以作为活性位点,大大提升镍基电极的催化性能[19,51]。采用电沉积法制备镍基自支撑电极的过程中,通过调控沉积条件就可制备出低结晶度或非晶态的催化功能层。Zai 等[19]在NF上直接沉积低结晶度的NiGaFe氢氧化物纳米片,研制了一种具有分级结构的镍基自支撑电极Ni0.65Ga0.30Fe0.05/NF。分级结构使电极具有优越的传质能力,低结晶度结构使其具有丰富的活性位点和较高的导电率,Fe、Ga 元素的引入优化了OER 过程中氧中间体的吸附能,从而促进了镍基电极OER性能的提升。Liu等[51]在NF上设计合成了银掺杂的氢氧化镍钴复合异质纳米材料[Ag@NiCo(OH)x],首先通过恒电位电沉积法合成NiCo-LDH 催化层,再向其中掺杂Ag 离子,研究发现掺杂Ag 后NiCo-LDH 催化层的晶体结构被严重破坏,产生非晶态结构。该电极在100mA/cm2的电流密度下,过电位比IrO2/NF电极减小了217mV,比NiCo/NF电极减小了42mV。这是由于Ag和NiCo(OH)x之间的电子转移以及非晶/晶体异质结构,使得电极兼具高电导性、丰富的活性位点和气体传输通道,同时通过循环伏安法证实电极具有高ECSA,具有丰富的活性位点和较高的本征活性,显著提升了Ag@NiCo(OH)x/NF电极的OER性能。
采用电沉积法除了合成双金属和三金属镍基自支撑电极外,四金属镍基自支撑电极也进入研究者的视野,不同金属元素之间的协同作用显著增强了电极的OER 反应活性,但在电沉积多金属元素掺杂的机理及活性位点的确定方面还需要进一步研究。Zhang 等[41]在镍网上电沉积生长了一种无黏结剂的NiFeWMo 合金纳米结构,并研究了该电极在常温和80℃下的电催化OER 性能。结果表明,在常温下,当电流密度为10mA/cm2时,过电位只需要152mV,在80℃条件下达到同样电流密度所需的过电位仅为75mV。经过6000 次循环测试后,LSV曲线均无明显变化,该四金属镍基电极在两个温度下都拥有良好的稳定性。通过在镍网上直接电沉积生长无黏结剂的合金纳米结构,使得电极具有较短的扩散路径,促进了合金的协同催化作用,从而提高了镍基电极的OER性能。
综上所述,采用电沉积方法,通过调节沉积电位或电流密度、沉积时间及沉积液类型,即可制备OER性能优异的双金属及多元金属镍基自支撑电极。通过精细调控催化功能层的结构、形貌和组成,改变了电极表面的原子构型,增强了金属之间的协同作用及活性位点的暴露,缩短了反应物和气体的扩散路径,进一步优化了OER 过程中中间体的吸附和活化过程,提升了镍基自支撑电极的OER性能。
2.3 非金属元素掺杂的镍基自支撑电极
过渡金属元素的掺杂虽然可以大大提升镍基自支撑电极的OER 性能,但在强氧化环境下的长期稳定性还有待提高。在镍基催化功能层中引入非金属元素(如磷、硫、氮等),形成非金属元素掺杂的镍基自支撑电极,不仅具有良好的OER 催化性能,而且具有更强的耐腐蚀能力[52-53]。非金属元素的掺杂不仅可以改善镍基自支撑电极催化功能层的纳米结构,增加活性位点数量,而且非金属负离子可以在OER 过程中有效调节金属的电子结构,从而提高活性位点的本征活性。
纳米结构过渡金属磷化物因其优异的导电性、良好的OER活性和稳定性而备受关注。Li等[54]通过一锅电沉积方法,在NF 上合成具有纳米片阵列结构的镍铁磷酸盐/磷化物(NiFe-Pi/P)催化功能层。电沉积工艺精细控制了NiFe-Pi/P 的纳米形貌,保证了材料丰富的活性位点数量和相结构的形成。材料具有杂化微晶粒嵌入的非晶结构,影响了催化剂表面原子构型,产生了较多的缺陷和空位,增大了活性位点,杂化微晶粒进一步稳定了结构,加倍提高了本征催化活性。材料也表现出超亲水性和超疏水性的特性,进一步提高了催化电极上的传质效率,使NiFe-Pi/P/NF拥有优越的OER性能。
镍基硫化物在OER 反应条件下可以逐渐转化为对应的氧化物,更容易形成羟基氧化物,进一步降低OER 过电位。Ni3S2催化剂由于含有丰富的Ni—S和Ni—Ni键,能诱导形成更具靶向性的OER中间体(HOO*),一直备受研究人员的关注[55-56]。基于以上研究,Xu 等[52]通过调节沉积电解液组成和pH,采用高温大电流电沉积技术,在NF上沉积了超薄Ni(OH)2/Ni3S2异质结构纳米片。松散的纳米结构有助于活性位点的充分暴露,并改善电解质渗透和气泡逸出行为,从而促进析氧反应动力学。此外,异质结构的相互协同作用同样促进了电解水反应的进行。Ni3S2电催化剂对OH-的吸附能力较弱,但Ni(OH)2助催化剂具有很强的OH-吸附能力,在OER 半反应中,H2O 分子可以在Ni(OH)2一侧有效地解离为OH-中间产物,促进反应的进行。Zhang等[57]通过梯度电沉积法,即在整个沉积过程中改变了4 次电流密度,得到了由NF 支撑的具有分枝状结构的NiCoS催化功能层,该结构和共掺杂优化的电子结构共同导致了ECSA迅速扩大,同时元素掺杂后金属和非金属之间的强协同效应也促进了OER反应的进行。
研究者也将多种非金属元素掺杂到镍基自支撑电极中,如图10所示,Khodabakhshi等[58]采用恒电流电沉积方法(10mA/cm2沉积30min),在含有NaH2PO4和Ni2+、Fe2+、Cu2+离子的盐溶液中,在Ni3S2/NF 衬底上沉积合成了具有高褶皱结构的三金属NiFeCu磷化异质结纳米片(NiFeCuP@Ni3S2/NF)。该电极在10mA/cm2的电流密度下,过电位为230mV,Tafel 斜率为42mV/dec,具有优异的OER 性能。这是由于非金属元素掺杂后形成的异质结纳米结构暴露了更多的活性位点,优化了相邻原子的电子结构并提高了电极的电导率。Xu 等[59]采用两步电沉积法直接在NF 上将NiS 和Ni2P 纳米颗粒嵌入非晶态Ni(OH)2纳米片中,制备了一种掺杂多元非金属元素的具有多孔结构的Ni-S-P 异质结纳米片,这些纳米片连接形成具有更大比表面积的花瓣状结构,为OER 反应提供了大量的活性位点和气体传输通道。此外,纳米片中的NiOOH、Ni(OH)2、Ni2P 和NiS组分原位生成了许多异质结,界面处的协同作用可以进一步促进了OER 反应的进行。Zhang 等[60]采用简易梯度电沉积法制备了S、P 两种非金属元素掺杂的镍基自支撑电极NiSP/NF,在NF 基底上合成了具有蜂巢状结构的催化功能层。由于梯度电沉积过程加速了电子转移,使得不同尺寸的Ni2P和NiS纳米颗粒聚集在基底表面形成了分层的纳米蜂窝结构,NiSP/NF 电极的ECSA 显著增大,暴露了更多的活性位点,同时由于Ni2P和NiS的良好的导电性以及相互之间的协同效应,使得NiSP/NF电极表现出优越的OER性能。

图10 NiFeCuP@Ni3S2/NiF三金属磷化物纳米片制造过程的示意图[58]
3 结语与展望
随着风光资源的大规模开发,将风能、光能转化为电能,进一步通过电解水制氢技术得到绿氢,将有效支撑未来氢能产业链的构建和绿氢的规模化利用。碱性电解水作为目前规模最大、商业化程度最高的电解水制氢技术,制氢效率仍有待提高。通过开发新型高活性析氧电极,降低阳极过电位,可以有效提高电解水制氢效率,降低制氢成本。本文简述了碱性电解水析氧原理和电沉积工艺,并综述了采用电沉积法制备镍基自支撑析氧电极的研究进展,包括在镍网或泡沫镍基底上制备镍(氢)氧化物、双金属及多元金属以及非金属掺杂的镍基催化剂。通过增强镍基催化剂的电导率及金属间的协同作用、增加活性位点数量、减小扩散路径以及改变表面原子构型等方式,可以有效改善镍基自支撑电极的OER 性能。电沉积方法具有条件温和、工艺简单、成本效益高、易于规模化制备等优势,是工业化生产镍基析氧电极的理想工艺,在碱性电解水领域中有广阔的应用前景。
尽管在基础研究工作中,电沉积法制备得到的镍基自支撑电极具有良好的OER 催化活性,但面向未来推广应用,仍需关注以下几个方面:①需要建立面向工业应用的电极材料标准评价体系,考虑如可再生能源电力输入波动性、长时间运行等实际工况,以在新型电极开发过程中客观评估其催化性能和应用潜力;②需要设计提高整个沉积系统的效率,提高沉积液浓度的均匀稳定性,以实现电沉积过程的可控高效,和放大制备过程中电极的均一稳定;③面向大规模生产与应用,需要开发完善的成套制备技术,包括电沉积溶液的回收利用和处理工艺,以实现电极的低成本清洁生产。
猜你喜欢
长江蔬菜(2021年22期)2022-01-12
食品安全导刊(2021年20期)2021-11-28
上海建材(2020年12期)2020-04-13
长江蔬菜(2018年22期)2018-12-25
长江蔬菜(2018年6期)2018-05-08
电镀与环保(2017年5期)2017-12-19
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
电镀与环保(2016年2期)2017-01-20
