
超高效液相色谱-串联质谱法测定化妆品中15种N-亚硝胺化合物
2023-11-29 01:43:20梁文耀何国山陈张好周智明席绍峰谭建华
分析测试学报 2023年11期
汪 毅,梁文耀,何国山,陈张好,周智明,吴 谦,席绍峰,谭建华*
(1.广州质量监督检测研究院,国家化妆品质量检验检测中心(广州),广东 广州 511447;2.广东省药品检验所,广东 广州 510663)
N-亚硝胺化合物是一类具有N-亚硝基结构的化合物,因取代基的不同,形成了种类繁多的同系物,目前已发现超过300种[1]。N-亚硝胺化合物大多具有致癌性[2],在饮用水、食品、烟草、化妆品中均有N-亚硝胺化合物检出情况[3-6],对人体健康构成威胁。我国《化妆品安全技术规范》(2015年版)[7]与欧盟化妆品法规(EC)No 1223/2009[8]均将N-亚硝胺化合物列入禁用目录,并规定化妆品产品中N-亚硝胺化合物不得超过50 ng/g。化妆品中N-亚硝胺化合物的主要来源为原料直接带入或原料经亚硝化反应产生。
文献报道化妆品中可能检出的N-亚硝胺化合物有N-亚硝基二乙醇胺(NDELA)、N-亚硝基二甲基胺(NDMA)等[6,9-14],检测方法主要有气相色谱-热能分析法(GC-TEA)[15]、液相色谱-热能分析法(LCTEA)[16]、液相色谱-串联质谱法(LC-MS/MS)[3,5,10,12-14]、气相色谱-串联质谱法(GC-MS/MS)[6,9,11,17]、液相色谱-高分辨质谱法(LC-HRMS)[4,18]、气相色谱-高分辨质谱法(GC-HRMS)[19]。N-亚硝胺化合物通常痕量存在于化妆品中,对方法的灵敏度要求极高。国家标准GB/T 29669-2013 采用GC-MS/MS 测定10 种N-亚硝胺化合物[20],但方法的检出限不能满足50 ng/g 的限量要求。另外,由于N-亚硝胺为一大类化合物,物化性质差异很大,现有文献采用LC-MS/MS 技术[3,5,10,12-14]进行测定时主要针对部分性质差异较小的N-亚硝胺化合物,方法所涉及的化合物覆盖范围不足。因此,亟需建立准确、灵敏、覆盖范围广的N-亚硝胺化合物检测方法。
本研究根据15种N-亚硝胺化合物的物化性质和不同化妆品的基质特点,通过系统优化前处理方法和仪器分析条件,最终采用分组提取,结合亚铁氰化钾-乙酸锌溶液沉淀大分子和饱和氯化钠-乙腈盐析等净化方法,解决了部分N-亚硝胺化合物稳定性差、易被干扰且多组分测定时存在的溶剂效应等问题,建立了覆盖化妆品中15种N-亚硝胺化合物的UPLC-MS/MS检测方法。方法的准确度、精密度和灵敏度可满足化妆品中N-亚硝胺化合物的痕量检测要求。
1 实验部分
1.1 仪器与试剂
SCIEX Triple Quad™ 5500+超高效液相色谱-三重四极杆质谱仪配有大气压化学电离源(APCI)(美国SCIX 公司);BSA224S-CW 电子天平(德国赛多利斯公司);MS3 basic 涡旋振荡器(德国IKA 公司);KQ-250DV型数控超声波清洗仪(昆山市超声仪器有限公司);Milli-Q纯水系统(美国Millipore公司)。
15 种N-亚硝胺化合物对照品和5 种N-亚硝胺化合物同位素内标信息详见表1,质量浓度均为100 μg/mL于甲醇,均购于上海安谱实验科技股份有限公司;甲醇、乙腈(色谱纯,德国Merck公司);甲酸(色谱纯,美国Sigma-Aldrich 公司);亚铁氰化钾、乙酸锌、乙酸铵、氯化钠(分析纯,广州化学试剂厂);实验用纯水(18.2 MΩ•cm)由Milli-Q纯水系统制备。化妆品的实际样品为市售产品。
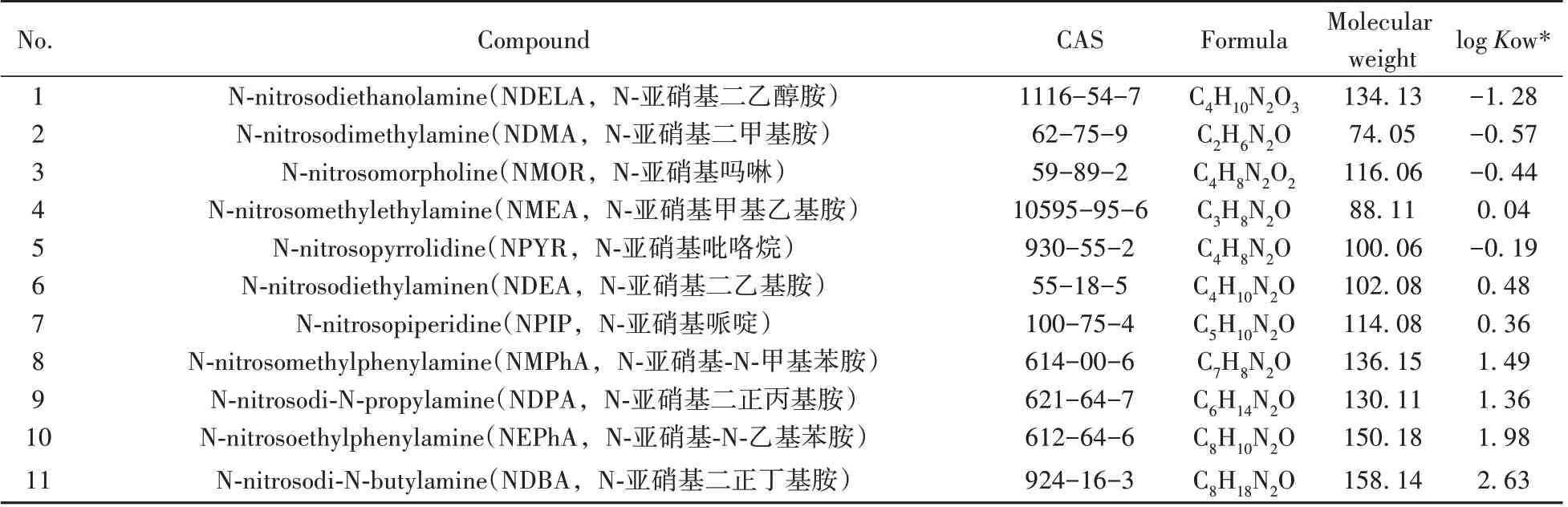
表1 N-亚硝胺化合物对照品和同位素内标信息Table 1 Reference materials and isotope internal standard compounds information of N-nitrosamine compounds

(续表1)
1.2 标准溶液的配制
分别准确移取NDELA、NDMA、NMOR、NMEA、NPYR、NDEA、NPIP、NDPA 对照品溶液各100 μL,置于10 mL棕色容量瓶中,用水定容,即得质量浓度为1 000 ng/mL的混合标准溶液(水);分别准确移取NDELA-D8、NDMA-D6、NPYR-D4、NDEA-D4内标溶液100 μL 于10 mL 棕色容量瓶中,用水定容,配制成质量浓度为1 000 ng/mL的混合内标溶液;用水配制成质量浓度为1、2、5、10、20、50 ng/mL,内标浓度为10 ng/mL 的标准工作溶液(水)。按相同的操作用乙腈配制NMPhA、NEPhA、NDBA、NDPhA、NDBzA、NDCH、NDiNA,内标为NDBA-D18的标准工作溶液(乙腈)。
1.3 样品前处理
1.3.1 水剂样品称取试样约0.5 g(精确至0.000 1 g)于10 mL 具塞比色管中,加入50 μL 混合内标溶液(水),混匀,用水定容至5 mL,超声提取10 min,经微孔滤膜过滤后,滤液作为待测溶液测定NDELA、NDMA、NMOR、NMEA、NPYR、NDEA、NPIP、NDPA。
称取试样约0.5 g(精确至0.000 1 g)于10 mL 具塞比色管中,加入50 μL 内标溶液(乙腈),混匀,用乙腈定容至5 mL,超声提取10 min,经微孔滤膜过滤后,滤液作为待测溶液测定NMPhA、NEPhA、NDBA、NDPhA、NDBzA、NDCH、NDiNA。
1.3.2 膏霜乳液样品称取试样约0.5 g(精确至0.000 1 g)于10 mL 具塞塑料离心管中,加入50 μL混合内标溶液(水),混匀,用水定容至5 mL,超声10 min,加入100 μL 2.5%亚铁氰化钾溶液和100 μL 5%乙酸锌溶液,混匀,静置5 min 后,以5 000 r/min 离心10 min,上清液经微孔滤膜过滤后,滤液作为待测溶液测定NDELA、NDMA、NMOR、NMEA、NPYR、NDEA、NPIP、NDPA。
称取试样约0.5 g(精确至0.000 1 g)于10 mL 塑料离心管中,加入50 μL 内标溶液(乙腈),混匀,加入2 mL 饱和氯化钠水溶液,混匀后,加入5 mL 乙腈,超声提取10 min,以5 000 r/min 离心10 min,上清液经微孔滤膜过滤后,滤液作为待测溶液测定NMPhA、NEPhA、NDBA、NDPhA、NDBzA、NDCH、NDiNA。
1.4 仪器条件
色谱柱:Agilent Poroshell 120 SB-Aq 色谱柱(100 mm×3.0 mm,2.7 μm,美国安捷伦公司);流动相:A为0.2 mmol/L乙酸铵水溶液(含0.1%甲酸),B为甲醇;梯度洗脱程序:0~2.0 min,2% B;2.0~10.0 min,2%~98% B;10.0~12.0 min,98% B;12.0~12.1 min,98%~2% B;12.1~15.0 min,2% B;流速:0.6 mL/min;柱温:40 ℃;进样体积:10 μL。电离方式:APCI+;气帘气:0.172 MPa;针电流:3 mA;源温度:450 ℃;喷雾气:0.207 MPa;喷撞气:0.062 1 MPa;扫描模式:多反应监测模式,15种亚硝胺化合物和5种同位素内标的质谱参数见表2。

表2 N-亚硝胺化合物和同位素内标的质谱参数Table 2 MS parameters of N-nitrosamine compounds and isotope internal standards
2 结果与讨论
2.1 质谱参数优化
电喷雾离子源(ESI)一般适用于分子量稍大、极性较强的化合物,大气压化学电离源(APCI)适用于分子量较小、极性稍弱的化合物。本研究的15种N-亚硝胺化合物的分子量和极性差异较大,导致其在ESI和APCI两种离子源的响应有较大差异。由于NDMA、NMEA、NDEA分子量较小且具有一定挥发性,这些化合物在APCI+模式下有较高的响应,但在ESI+模式下响应非常低。对于NDBA、NDPhA、NDBzA、NDCH、NDiNA 等化合物,由于支链的碳链不断加长,分子量变大,在APCI+模式下响应强度低于ESI+,但仍然能够满足灵敏度要求。进一步分析基质加标样品时发现,膏霜乳液类化妆品在ESI+下相比APCI+更易出现基质干扰和抑制,特别是对于出峰时间较慢的NDBA、NDPhA、NDBzA、NDCH、NDiNA 等化合物,其原因可能是因为化妆品中各类高含量表面活性剂等基质所致。因此,本研究选择APCI+模式,并通过进一步优化源温度、喷雾气、针电流参数使15 种N-亚硝胺化合物获得最高的仪器灵敏度。
2.2 色谱条件优化
2.2.1 色谱柱的选择考察了Thermo Hypersil GOLD C18色谱柱(50 mm×2.1 mm,1.9 μm,美国赛默飞公司)、Agilent Poroshell Bonus-RP(50 mm×4.6 mm,2.7 μm,美国安捷伦公司)、Agilent Poroshell SB-C18色谱柱(100 mm×2.1 mm,2.7 μm,美国安捷伦公司)、Phenomenex Kinetex C18色谱柱(100 mm×3 mm,2.6 μm,美国Phenomenex 公司)、Agilent Poroshell SB-Aq 色谱柱(100 mm×3 mm,2.7 μm,美国安捷伦公司)5 种色谱柱对N-亚硝胺化合物的分离情况。结果显示,化合物在5 种色谱柱上均能实现分离,柱径和填料粒径越小,峰形越尖锐,但柱压越高,其中Aq柱对亲水性化合物保留较好,能使化合物与基质干扰分离。考虑到APCI 需要较高流速,且分离NDELA 和NDMA 需要高比例水相,同时为了减少化妆品基质成分的干扰,最终选用Agilent Poroshell SB-Aq 色谱柱进行分离。
2.2.2 流动相的选择考察了水+甲醇(A)、0.1%甲酸水+甲醇(B)、0.1%甲酸水+0.1%甲酸甲醇(C)、1 mmol/L 乙酸铵水溶液(含0.1%甲酸)+甲醇(D)、1 mmol/L 乙酸铵水溶液(含0.1%甲酸)+乙腈(E)5 组不同流动相对化合物峰形和响应的影响。结果显示,使用流动相A、B、C 时,15 种N-亚硝胺化合物均能正常出峰,但NDMA、NMEA 和NPYR 检测离子对的基线响应较高,信噪比偏低,灵敏度下降。甲酸的加入能使化合物响应增大,但对基线响应偏高的现象无改善,且多个化合物的基线响应波动增大。使用流动相D 时,乙酸铵的加入明显降低基线响应和改善峰形,但同时也会抑制部分化合物的电离,降低响应,但总体来说提高了信噪比,特别是提高了NDMA、NMEA 和NPYR 的信噪比。流动相E 用乙腈代替甲醇作为有机相,峰形变得更尖锐,但所有化合物的响应明显降低。分析其原因可能是在APCI+模式下,甲酸的加入能提供H+质子,有助于化合物电离的同时也提高了背景化合物的响应。乙酸铵的加入,使得流动相离子强度增加,电子转移速率增加,目标化合物和背景化合物同时抑制电离,乙酸铵在一定浓度下,可提高目标化合物的信噪比。
在流动相D 的基础上进一步对比了不同乙酸铵浓度(0.1、0.2、0.5、1、2 mmol/L)对化合物峰形及响应的影响。结果显示,随着乙酸铵浓度的增大,基线响应降低,0.5 mmol/L时基线响应达到最低,继续增大乙酸铵浓度,对基线影响不明显。化合物响应则随乙酸铵浓度的增大而降低,NMOR 较为明显。为了兼顾基线响应和化合物响应,达到最优信噪比,最终选取0.2 mmol/L 乙酸铵水溶液(含0.1%甲酸)+甲醇作为流动相。优化条件下15 种N-亚硝胺化合物和5 种同位素内标的提取离子流色谱图见图1。
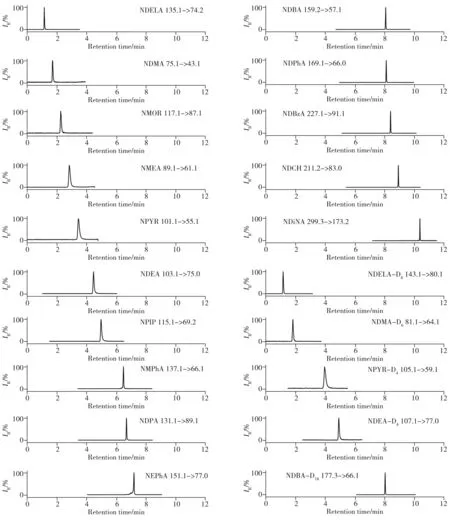
图1 15种N-亚硝胺化合物和5种同位素内标提取离子色谱图Fig.1 Extraction ion chromatograms of 15 N-nitrosamine compounds and 5 isotope internal standards
2.3 前处理条件的优化
2.3.1 提取方式的优化15 种N-亚硝胺化合物的性质差异较大,logKow 从-1.28 到7.07,可溶于甲醇、乙腈等有机溶剂且部分可溶于水。实验首先考察了不同上机溶液(纯水、10%~90%甲醇水、10%~90%乙腈水、甲醇和乙腈)对溶剂效应的影响。结果显示,NDELA、NDMA、NMOR、NMEA、NPYR、NDEA、NPIP、NDPA 这8种脂溶性较低的N-亚硝胺化合物对上机溶液较为敏感,溶剂效应较强,有机相比例增大时,峰形严重展宽。同时,随着有机相比例增大,脂溶性较强的NDiNA 响应逐步提高,原因可能是由于有机相比例提高,增大了溶解度,使其响应提高(见图2)。因此,由于溶剂效应和溶解度的双重影响,本研究中的15种N-亚硝胺化合物很难选取同一种上机溶液。

图2 不同上机溶液中N-亚硝胺化合物的提取离子色谱图Fig.2 Extraction ion chromatograms of N-nitrosamine compounds in different solutions
为避免溶剂效应,实验进一步以乙腈为提取溶剂,考察增加氮吹、复溶的方式对N-亚硝胺化合物加标回收率的影响。结果显示,15 种化合物的加标回收率均小于80%,分子量较小的NDMA、NMEA、NDEA等的回收率甚至低于50%。进一步研究发现,即使严格控制温度、避光和氮气流量条件,氮吹过程中N-亚硝胺化合物仍有较大损失,说明该类化合物的稳定性较差。因此整个实验过程应避免使用氮吹处理。如果采用纯水对提取液稀释的方式降低溶剂效应,上机溶液中乙腈的比例需稀释至低于20%,但该操作将方法检出限提高了5倍,无法满足50 ng/g的法规限量要求。因此,本研究将15种N-亚硝胺化合物根据极性大小分为两组,第一组为NDELA、NDMA、NMOR、NMEA、NPYR、NDEA、NPIP、NDPA,第二组为NMPhA、NEPhA、NDBA、NDPhA、NDBzA、NDCH、NDiNA,分别用水和乙腈进行提取。
进一步实验发现,对于第一组化合物采用水作为提取溶剂,膏霜、乳液样品含较多脂质和增稠剂,提取后难于过滤,水剂基质则能很好实现提取和过滤。而在膏霜、乳液基质提取液中加入亚铁氰化钾-乙酸锌溶液沉淀大分子,离心后,可顺利完成过滤操作。该条件下,NDELA 等8 种N-亚硝胺化合物的加标回收率均大于85%,且上机测定时色谱峰形尖锐。对于第二组化合物直接采用乙腈进行提取,但部分膏霜样品因含有高含量的表面活性剂等,造成较为严重的基质干扰。采用饱和氯化钠-乙腈盐析萃取法能大大降低基质效应,NMPhA等7种N-亚硝胺化合物的加标回收率均在85%以上。
2.3.2 提取时间的优化选取超声提取方式进行样品的前处理,并考察了不同超声时间(5、10、15、20 min)对化合物的提取效果(见图3)。结果显示,超声时间达到10 min 时,15 种N-亚硝胺化合物的加标回收率均可达到85%以上,进一步延长超声时间对提取效果影响不大,但在超声时间达到20 min 时部分N-亚硝胺的回收率呈下降趋势,可能是发生了降解转化所致。因此本研究选取10 min 作为最佳超声提取时间。

图3 超声时间对15种N-亚硝胺化合物回收率的影响Fig.3 Effect of ultrasound time on the spiked recoveries of 15 N-nitrosamine compounds
2.3.3 固相萃取净化效果的考察采用固相萃取的净化方式考察了Waters Oasis HLB(60 mg,3 mL)、Waters Oasis WCX(60 mg,3 mL)、Waters Oasis MCX(60 mg,3 mL)(美国沃特世公司)和CNW C18(60 mg,3 mL,上海安谱公司)4 种萃取小柱对N-亚硝胺化合物的加标回收率。结果显示,使用HLB 和C18小柱时,过柱和氮吹等操作导致部分化合物损失严重,且强极性的NDELA和NDMA在固相萃取小柱上保留较弱,导致直接流出,回收率低于10%;而NDiNA 在小柱上保留过强,难于洗脱,使得回收率偏低。使用WCX 和MCX 时,上样和洗脱需用氨水和甲酸调节pH 值,由于N-亚硝胺化合物对酸碱较敏感,操作过程中导致化合物降解,回收率无法达到理想效果。因此,本研究最终未采用固相萃取的净化方式,而是通过“2.3.1”对提取方式进行系统优化以提升方法的准确性和精密度。
2.4 线性关系、方法检出限与定量下限
按“1.4”仪器条件,将质量浓度为1、2、5、10、20、50 ng/mL,内标浓度为10 ng/mL 的标准工作溶液由低到高进行测定。以标准溶液浓度与同位素内标浓度之比为横坐标,对应的峰面积与同位素内标峰面积之比为纵坐标,绘制标准曲线。得到线性回归方程和相关系数,所有化合物在相应质量浓度范围内均呈现良好线性,相关系数(r2)均大于0.995。分别以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)计算方法检出限(LOD)和定量下限(LOQ)。表3 列出了15 种N-亚硝胺化合物的线性方程、相关系数、方法检出限和定量下限。
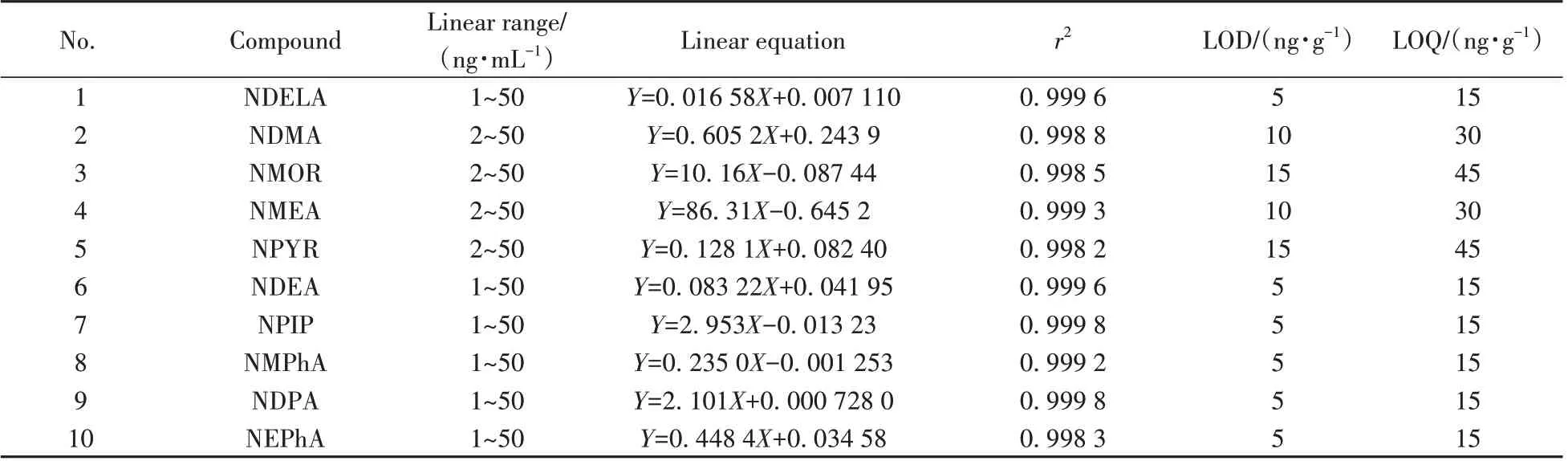
表3 15种N-亚硝胺化合物的线性回归方程、相关系数(r2)、方法检出限及定量下限Table 3 Linear regression equations,correlation coefficients(r2),detection limits and quantitation limits of 15 N-nitrosamine compounds

(续表3)
2.5 回收率与相对标准偏差
平行称取3 种空白基质(水剂、乳液、膏霜)样品各6 份,每份约0.5 g,共3 组,分别加入适量混合标准溶液和内标溶液,使得加标水平分别为25、50、100 ng/g(1/2倍限量值、限量值、2倍限量值),按“1.3”处理后进行测定,15种N-亚硝胺化合物的加标回收率与相对标准偏差(RSD)结果见表4。从表中可以看出,3 种基质在不同加标浓度下化合物的平均回收率为88.0%~111%,RSD 为1.4%~9.8%。结果显示,该方法准确可靠,精密度高。
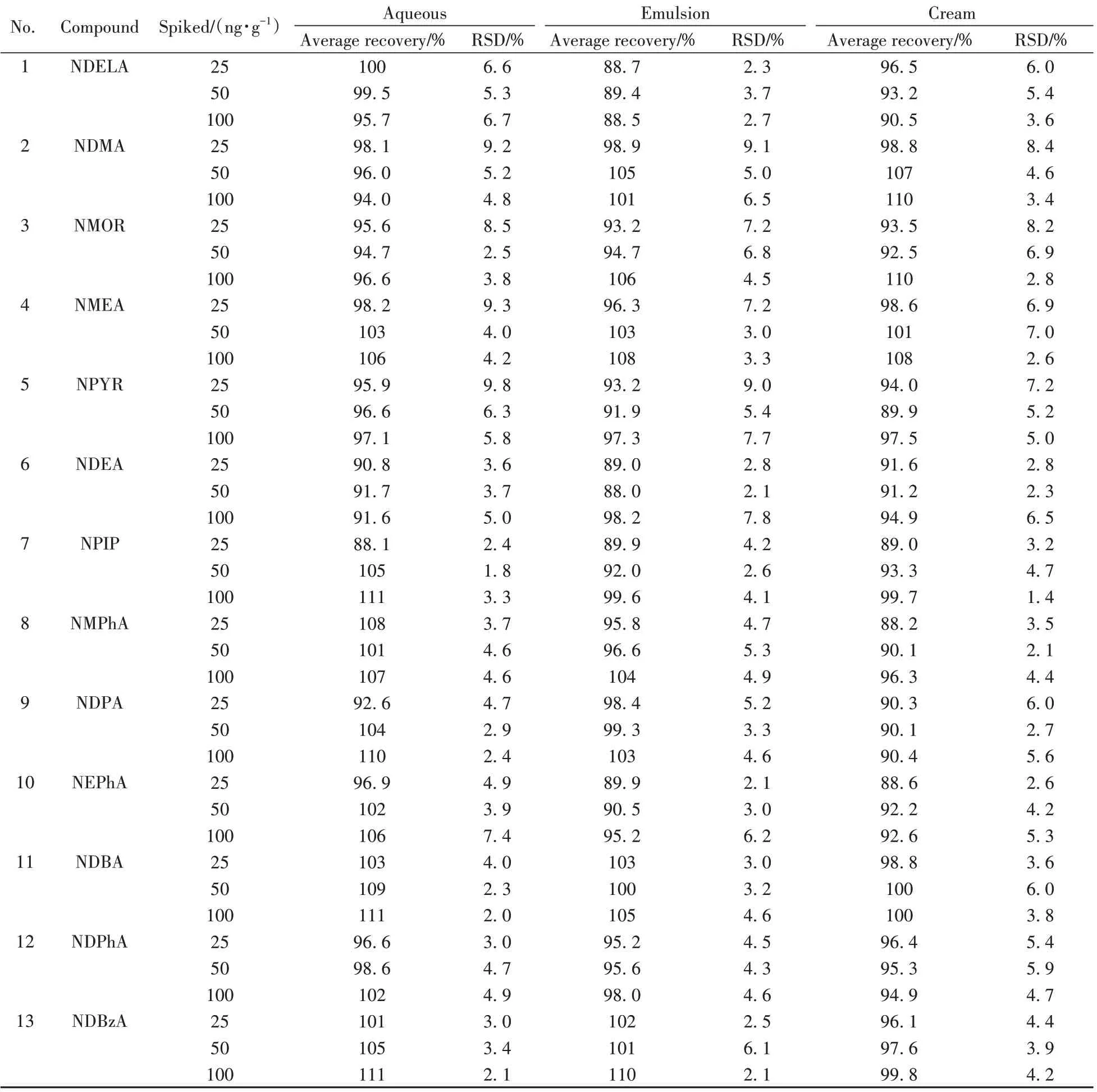
表4 15种N-亚硝胺化合物的加标回收率及相对标准偏差(n=6)Table 4 Spiked recoveries and RSDs of 15 N-nitrosamine compounds(n=6)
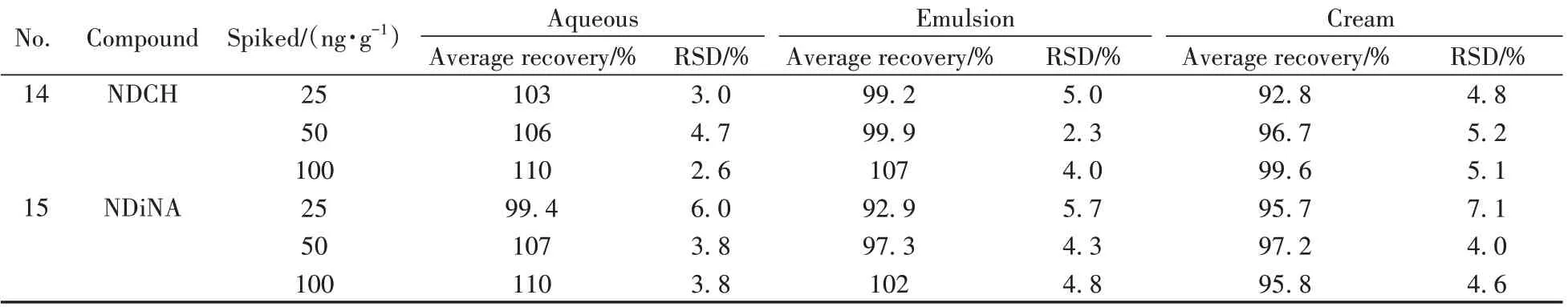
(续表4)
2.6 实际样品测定
收集水剂、乳液、膏霜等类型市售化妆品样品71 批次,采用本方法对15 种N-亚硝胺化合物进行测定。检测结果见表5。13 批次样品检出NDELA,其中12 批次均小于限量值50 ng/g,1 批次洁面泡泡样品中检出705 ng/g的NDELA,超出限量值。化妆品样品中常检出NDELA,这与化妆品较多使用三乙醇胺(TEA)相关原料有关。分析NDELA 超限量值样品的配方表,发现其使用月桂醇硫酸酯TEA盐作为原料,此原料可能带入NDELA,导致其超过限量值,建议加强原料的质量控制。
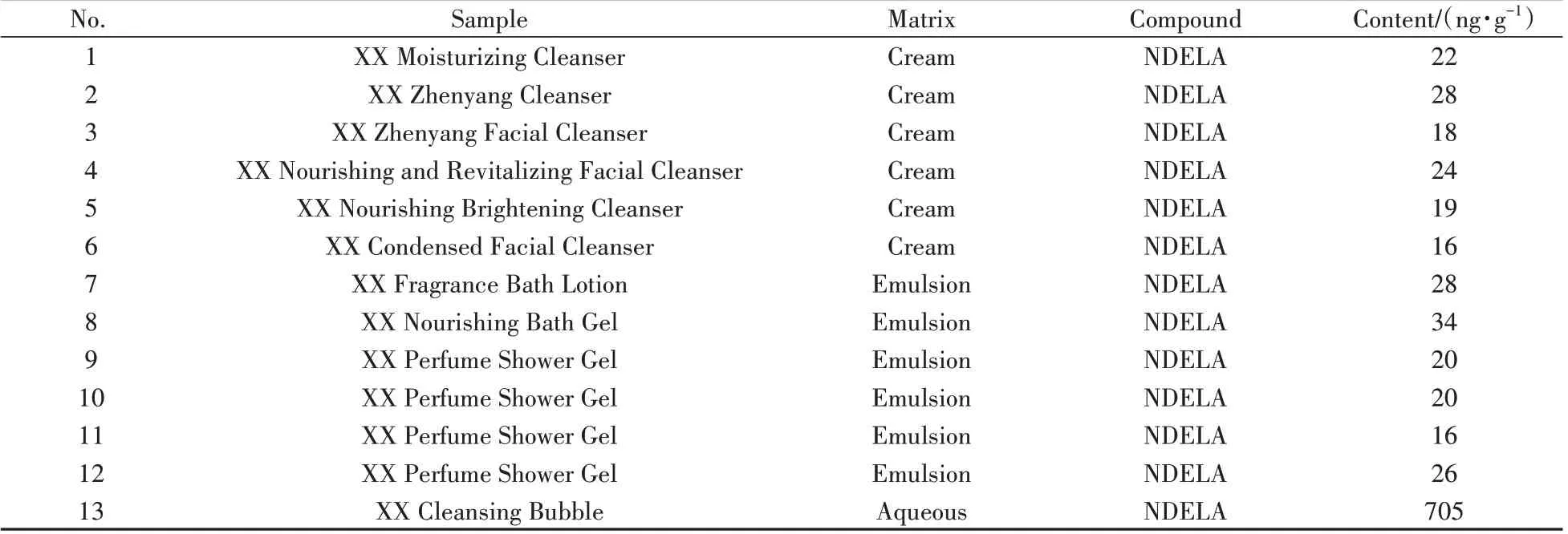
表5 市售样品中N-亚硝胺化合物的检测结果Table 5 Test results of N-nitrosamine compounds in commercially available samples
3 结 论
本研究通过优化质谱参数和超高效液相色谱条件,调整前处理方法以及采用同位素内标等方式,建立了UPLC-MS/MS测定化妆品中15种N-亚硝胺化合物的分析方法。方法针对性较强、测定结果准确可靠,检出限满足相关限量要求,适用于化妆品中15种N-亚硝胺化合物的痕量测定。该方法有效弥补了现有标准方法的不足,通过对市售化妆品的检测,发现NDELA常检出且存在超限量值情况。本方法的建立将为化妆品中N-亚硝胺化合物的监测和风险评估提供更多的技术手段,为化妆品监管和产品质量控制提供重要的技术支撑。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
云南化工(2020年11期)2021-01-14 00:50:38
食品安全导刊(2020年21期)2020-09-07 09:14:04
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
中成药(2018年6期)2018-07-11 03:01:32
中成药(2017年10期)2017-11-16 00:50:29
山东工业技术(2016年13期)2016-06-29 09:05:13
中国粮油学报(2016年5期)2016-01-23 02:45:06
