
超高效液相色谱-串联质谱法测定化妆品中全氟及多氟化合物
2023-11-29 01:42:52梁梓洋张秋炎康怀腾梁韵蕊轩申鑫吴惠勤陈桂琴李杨杰罗辉泰
分析测试学报 2023年11期
梁梓洋,张秋炎,周 熙,康怀腾,梁韵蕊,轩申鑫,吴惠勤,黄 芳,陈桂琴,李杨杰*,罗辉泰*
(1.广东省科学院测试分析研究所(中国广州分析测试中心),广东省化学测量与应急检测技术重点实验室,广东省中药质量安全工程技术研究中心,广东 广州 510070;2.广东省药品检验所,国家药品监督管理局化妆品风险评估重点实验室,广东 广州 510070)
全氟及多氟烷基化合物(PFASs)是一类分子中含有全氟或多氟烷基的化合物,具有较好的稳定性、抗降解性和疏水疏油性,因此自20世纪50年代起便被应用于生产具有耐污、防水、抗油等特性的日用品,在化工、纺织品、炊具等领域有着广泛的应用[1]。尽管《斯德哥尔摩公约》已将全氟辛酸(PFOA)、全氟辛磺酸(PFOS)及其盐类列入持久性有机污染物(Persistent organic pollutants,POPs)名单中,禁止或限制上述化合物的使用,但仍存在使用其它全氟化合物作为替代品的情况。因此,近年来越来越多的研究发现,PFASs 在空气、饮用水、食品中均有检出[2-3],广泛分布于环境当中[4],并且可以通过呼吸、饮食等多种途径进入人体,主要分布并积累在血液、肝脏和肾脏中[5]。毒理学研究表明,PFASs具有生殖毒性、诱变毒性、发育毒性、神经毒性、免疫毒性等多种毒性[6],是一类具有全身多脏器毒性的环境污染物,可引发包括癌症在内的多种不容忽视的健康危害,因此全球已有多个法规将PFASs 列入了禁止或限制使用清单,如欧盟在2019 年发布的(EU)2019/1021 中规定了几种PFASs 可允许的最大含量[7];美国国会在2021 年提出了禁止在食品包装中使用任何PFASs 的法案[8];而我国也在2023 年发布的《重点管控新污染物清单(2023 年版)》中明确禁止生产、加工和使用特定几种PFASs 及其盐类[9]。由此可见各国对PFASs的问题均给予了高度重视,对其使用具有严格的要求。
鉴于PFASs 在环境中的广泛分布及历史上的大量应用,对于PFASs 的研究多集中在环境、食品、纺织品等领域。然而,近年来多个研究发现化妆品中普遍存在PFASs[10-11],作为一种良好的疏水添加剂,PFASs阳性样品主要集中在乳液、口红、睫毛膏、粉底液等多种具有一定防水效果的化妆品中[12],检出的PFASs 浓度分布范围较广,最高可达19 μg/g[13]。对于消费者而言,若使用了含有PFASs 的化妆品,则可能通过皮肤或泪道被吸收,也可能通过鼻口吸入摄入,若长期使用将给使用者带来极大的安全健康隐患[14]。
目前国内外有关化妆品中PFASs检测方法的研究相对较少,仅针对某几种特定的PFASs[15-16],缺乏对不同种类化妆品中多种PFASs同时测定的方法研究,无法为后续研究提供方法基础。尽管在2021年6月,美国国会出台了“化妆品中不含PFASs”的法案,拟禁止在化妆品中故意添加PFASs,但目前仍缺乏具体的相关标准。而国内与PFASs 相关的现行标准涉及的领域多为纺织品、环境、食品等,针对化妆品的相关标准较少且PFASs 种类的涵盖面较窄,如SN/T 2393-2009[17]和SN/T 3694.1-2014[18]两个行业标准仅包含了全氟羧酸类化合物(PFCAs)和全氟磺酸类化合物(PFSAs)共10 个,品种覆盖面难以对国内化妆品市场中的PFASs 进行有效监管与控制。综合来看,有需要建立一种快速、简便、可靠的化妆品中多种PFASs的检测方法,以降低或控制化妆品的使用风险。
近年来,随着PFASs 的风险问题逐渐得到关注,相关检测方法的研究也越来越多,其中PFASs 的测定主要有液相色谱-串联质谱法(LC-MS/MS)[19-20]、气相色谱-串联质谱法(GC-MS/MS)[21-22]和气相色谱-质谱法(GC-MS)[23]。由于大部分目标PFASs 的沸点较高,采用GC-MS 分析时需要进行衍生化,增加了样品前处理步骤的复杂性,使用上有一定的限制,相比之下,LC-MS/MS 法可对大部分PFASs 直接测定,更适合相关的分析。
本文旨在建立化妆品中PFASs 的LC-MS/MS 分析方法,针对化妆品基质复杂的特点,通过优化液相色谱与质谱条件,建立专属性强和灵敏度高的仪器方法;通过优化操作,获得简便、准确度高、精密度好的前处理方法。本文所建立的方法适用于多种类型化妆品中PFASs 的快速检测,可为保障化妆品质量安全和我国化妆品高质量发展提供技术支撑。
1 实验部分
1.1 仪器与试剂
液相色谱-串联质谱仪(1290 Infinity Ⅱ/6470 Triple Quad,美国Agilent 公司);电子分析天平(美国Sartorious 公司);涡旋振荡器(德国IKA公司);0.22 μm有机微孔滤膜(美国Agilent公司)。
全氟丙酸(PFPrA,98.0%)、全氟十三酸(PFTriDA,91.9%)、全氟十六酸(PFHxDA,85.2%)、N-甲基全氟-1-辛烷磺酰胺基乙酸(NMeFOSAA,83.8%)、N-乙基全氟-1-辛烷磺酰胺基乙酸(NEtFOSAA,99.4%),购自广州佳途科技股份有限公司;全氟丁酸(PFBA,98.4%)、全氟戊酸(PFPeA,98.8%)、全氟己酸(PFHxA,99.3%)、全氟庚酸(PFHpA,99.5%)、全氟辛酸(PFOA,99.0%)、全氟壬酸(PFNA,97.1%)、全氟癸酸(PFDA,99.9%)、全氟十一酸(PFUnDA,98.2%)、全氟十二酸(PFDoDA,92.3%)、全氟庚基磺酸(PFHpS,96.2%),购自北京坛墨质检科技有限公司;全氟戊基磺酸(PFPeS,92.6%)、全氟己基磺酸(PFHxS,95.0%)、全氟辛基磺酸(PFOS,97.4%)、1H,1H,2H,2H-全氟己二磺酸(4∶2 FTSA,94.3%)、1H,1H,2H,2H-全氟辛基磺酸(6∶2 FTSA,94.4%)、全氟辛基磺酰胺(FOSA,97.0%)、N-甲基全氟辛基磺酰胺(MeFOSA,97.3%)、N-乙基全氟辛基磺酰胺(EtFOSA,98.07%)、2H,2H,3H,3H-全氟己酸(3∶3 FTCA,99.00%),购自德国Dr.Ehrenstorfer GmbH 公司;全氟十四酸(PFTeDA,99.0%)购自北京曼哈格生物科技有限公司;全氟丁基磺酸(PFBS,98%)购自上海毕得医药科技有限公司;2H,2H,3H,3H-全氟辛酸(5∶3 FTCA,1 mg/mL)、全氟(2-乙氧基乙烷)磺酸(PFEESA,1 mg/mL)、全氟-3-甲氧基丙酸(PFMPA,1 mg/mL)、全氟-4-甲氧基丁酸(PFMBA,1 mg/mL),购自天津阿尔塔科技有限公司;甲醇、乙腈、甲酸(色谱纯,德国Merck公司);超纯水(中国屈臣氏集团有限公司);氯化钠(分析纯,广州化学试剂厂);乙酸铵(分析纯,上海麦克林生化科技有限公司);88个实际样品来自市售化妆品。
1.2 对照品溶液的配制
储备液:精密称取各PFASs对照品,用甲醇配制成质量浓度为1 mg/mL 的对照品储备液,于4℃下保存。
中间液:用乙腈配制PFHxDA、4∶2 FTSA、6∶2 FTSA、FOSA、3∶3 FTCA、5∶3 FTCA、NEtFOSAA及NMeFOSAA的质量浓度为50 mg/L、其他22种PFASs为10 mg/L的混合标准储备溶液。
工作液:用空白基质液将中间液稀释成系列的基质匹配混合对照品工作液,其中PFHxDA、4∶2 FTSA、6∶2 FTSA、FOSA、3∶3 FTCA、5∶3 FTCA、NEtFOSAA 及NMeFOSAA 的系列质量浓度分别为5、10、25、50、100、250 μg/L,其他22种PFASs的系列质量浓度分别为1、2、5、10、20、50 μg/L。
空白基质液:称取经检测不含PFASs 的空白样品,按照“1.3”方法进行处理,得到空白基质溶液。
饱和乙酸铵-饱和氯化钠溶液(含1%甲酸):称取氯化钠45 g 于锥形瓶中,加100 mL 水后超声15 min,待其不再溶解后过滤,滤液即为饱和氯化钠溶液。称取乙酸铵160 g 于锥形瓶中,加入饱和氯化钠溶液100 mL 后超声15 min,待其不再溶解后过滤,滤液即为饱和乙酸铵-饱和氯化钠溶液。移取甲酸1 mL于100 mL容量瓶中,以饱和乙酸铵-饱和氯化钠溶液稀释并定容至刻度线,摇匀后即得。
饱和乙酸铵-乙腈溶液(含0.001%氨水):称取过量乙酸铵,加入乙腈后超声15 min,待其不再溶解后过滤,滤液即为饱和乙酸铵-乙腈溶液。移取20 μL 25%氨水于500 mL 容量瓶中,以饱和乙酸铵-乙腈溶液稀释并定容至刻度线,摇匀后即得。
1.3 样品前处理
对于水剂、膏霜、乳液、凝胶及粉类样品,称取样品0.2 g(精确至0.000 1 g)于50 mL 离心管中,加入饱和乙酸铵-饱和氯化钠溶液(含1%甲酸)1 mL,涡旋30 s 使之分散,随后精密加入2.5%甲酸-乙腈溶液10 mL,涡旋5 min后以8 000 r/min离心,取上层清液经0.22 μm有机滤膜过滤后待分析。
对于油状及蜡基样品,称取样品0.2 g(精确至0.001 g)于50 mL 离心管中,加入正己烷1 mL,涡旋30 s使样品分散,加入饱和乙酸铵-乙腈溶液(含0.001%氨水)10 mL,涡旋5 min,4 000 r/min 离心5 min,取下层清液经0.22 μm有机滤膜过滤后待分析。
1.4 仪器条件
1.4.1 液相色谱条件色谱柱:Agilent RRHD Eclipse Plus Zorbax C18色谱柱(3.0 mm × 100 mm,1.8 μm);捕集柱:EC-C18(2.1 mm × 50 mm,1.9 μm);流动相:A 为5 mmol/L 乙酸铵水溶液,B 为乙腈;洗脱程序:0~1 min,10% B;1~3 min,10%~30% B;3~5 min,30%~45% B;5~10 min,45%~70% B;10~15 min,70%~95% B;15~17 min,95% B;流速:0.35 mL/min;柱温:40 ℃;进样量:1 μL。
1.4.2 质谱条件离子源:电喷雾电离源(ESI),负离子模式;扫描模式:动态多反应监测(dMRM);毛细管温度:250 ℃;氮气流速:8 L/min;雾化气压力:45 psi;毛细管电压:2 500 V;鞘气温度:350 ℃;鞘气流速:12 L/min;喷嘴电压:1 000 V。30 种PFASs 的定性、定量离子对和碰撞能量(CE)等质谱参数如表1所示。
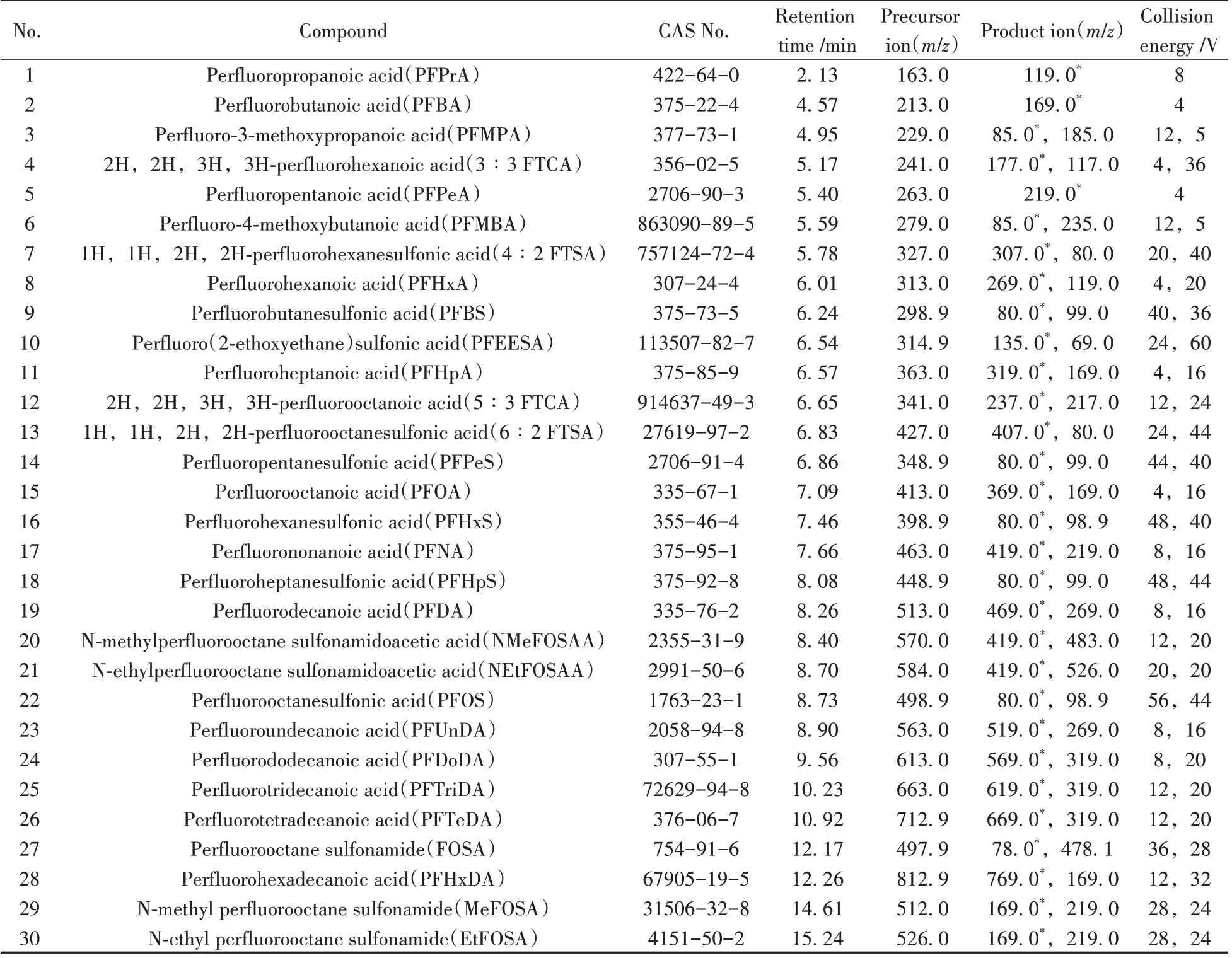
表1 30种PFASs的质谱参数Table 1 Mass parameters of 30 PFASs
2 结果与讨论
2.1 色谱条件的选择
取一定浓度的30 种PFASs 对照品溶液,分别用YMC-Triart C18(2.1 mm×100 mm,3.0 μm)、YMCTriart PFP(2.1 mm×100 mm,3.0 μm)、Agilent HPH-C18(2.1 mm×100 mm,2.7 μm)、SB-C18(2.1 mm×100 mm,2.7 μm)、Zorbax C18(3.0 mm×100 mm,1.8 μm)、HSS-T3(2.1 mm×100 mm,3.5 μm)、SBAq(2.1 mm×100 mm,2.7 μm)、EC-C18(2.1 mm×100 mm,2.7 μm) 8 种色谱柱进行分析,考察30 种PFASs 在不同色谱柱上的色谱行为。结果表明,在EC-C18和HPH-C18柱上,化合物普遍峰形差且响应低;在YMC-Triart PFP、SB-Aq、SB-C18和YMC-Triart C18柱上,峰形正常,但部分化合物不出峰,而使用Zorbax C18柱时各化合物具有较满意的峰形与响应,因此选择Zorbax C18柱进行后续分析。
分别选用水-乙腈、0.2%甲酸-乙腈、0.2% 乙酸-乙腈、2 mmol/L 乙酸铵-乙腈、5 mmol/L 乙酸铵-乙腈、10 mmol/L 乙酸铵-乙腈、2 mmol/L 乙酸铵(含0.2%甲酸)-乙腈和2 mmol/L 乙酸铵(含0.2%乙酸)-乙腈8种流动相,考察不同流动相体系对30种PFASs色谱行为的影响。结果表明,当使用水-乙腈体系时,化合物峰形与响应均较差;当使用0.1% 甲酸-乙腈和0.2% 乙酸-乙腈时,化合物峰形不佳;当使用2 mmol/L 乙酸铵-乙腈、5 mmol/L 乙酸铵-乙腈和10 mmol/L 乙酸铵-乙腈时,化合物峰形与响应均较好,且不同乙酸铵浓度的体系之间无明显差异;当在乙酸铵-乙腈体系中加入0.2%甲酸或0.2%乙酸时,较高的质子浓度会抑制[M-H]-离子的形成,使得化合物响应有所下降。综上考虑,故选择5 mmol/L 乙酸铵-乙腈体系作为最佳流动相,所得30 种PFASs的总离子流图如图1所示。

图1 30种PFASs的总离子流图Fig.1 Total ion chromatograms of 30 PFASs the numbers denoted were the same as that in Table 1
2.2 质谱条件的选择
取一定质量浓度的30种PFASs对照品溶液,选取响应较高的离子对,优化碰撞能量,得到定量离子对和定性离子对,具体的质谱参数见表1。
2.3 前处理条件的优化
化妆品种类多,常见的剂型为水剂、膏霜、乳液、凝胶、粉状、油状和蜡基,大部分基质具有较高的复杂性,为了降低化妆品基质对仪器的污染及测试结果的干扰,需对样品进行合适的前处理。
针对水剂、膏霜、乳液、凝胶和粉状样品,分别采用以下4 种方法进行提取:M1 为乙腈10 mL 超声提取;M2 为乙腈(含1%甲酸)10 mL 超声提取;M3 为以饱和乙酸铵-氯化钠溶液(含1%甲酸)1 mL 分散样品,再加入乙腈(含1%甲酸)10 mL 提取;M4 为以饱和乙酸铵-氯化钠溶液(含1%甲酸)2 mL 分散样品,再加入乙腈(含1%甲酸)10 mL 提取。针对油状和蜡基样品,分别采用以下两种方法进行提取:M5 为加入正己烷1 mL 分散样品,再加入饱和乙酸铵-乙腈溶液10 mL 提取;M6 为加入正己烷1 mL 分散样品,再加入饱和乙酸铵-乙腈(含0.001%氨水)10 mL 提取。使用上述方法对相应的样品进行前处理,按“1.4”仪器条件进行分析,考察不同前处理方法对不同剂型化妆品样品中30 种PFASs 回收率的影响,结果如图2所示。
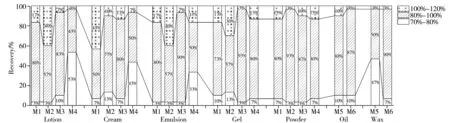
图2 不同前处理方法下,不同化妆品样品中30种PFASs的回收率Fig.2 Recoveries of 30 PFASs in different cosmetics with different pretreatments
对于水剂、膏霜、乳液、凝胶和粉状样品,考察的4种方法均可获得70%~120%的回收率,但考虑到化妆品基质的复杂性,若采用直接提取法(M1 和M2)将会提取到大量基质而有污染仪器的风险。若采用分散后液液萃取(M3 和M4),在甲酸作用下,部分带有羧基的PFASs 能够以酸形式存在,从而提高乙腈的萃取效率,同时饱和盐溶液可促进水相与乙腈的分层,使部分基质能够留在水相,对提取液起到一定的净化效果,从而减轻样品基质对仪器的污染。当用于分散样品的盐溶液体积由1 mL(M3)增至2 mL(M4)时,PFPrA、PFBA等极性较强的PFASs,容易进入水相而使得乙腈的萃取效率下降,回收率偏低。综上所述,当采用M3 时,80%以上的PFASs 能够获得80%~120%的回收率,故选择该方法作为水剂、膏霜、乳液、凝胶和粉状样品的前处理方式。
对于油状和蜡基样品,所考察的两种方法的回收率均在70%~120%范围内。由于PFASs 结构上呈疏水性,在同样为疏水性的油状和蜡基样品中有一定的溶解度,因此通过在乙腈中加入氨水和乙酸铵(M6),使带有羧基的PFASs以铵盐形式存在,降低其在正己烷中的溶解度,可在提高乙腈萃取效果的同时,使脂溶性基质留在正己烷层,从而起到部分净化的作用。综上,采用M6时87%以上的PFASs可获得80%~120%的回收率,故选择该方法作为油状和蜡基样品的前处理方式。
2.4 基质效应
在液相色谱-质谱分析中基质效应(Matrix effect,ME)普遍存在,在基质作用下待测物会发生电离抑制或电离增强,表现为检测信号减弱或增强,且待测物浓度较低时愈加明显,对定量结果的准确性和重复性有着严重的影响[24]。有多种方法可消除基质效应,如使用固相萃取(SPE)等净化样品,使用同位素内标对待测物的响应进行校正,或使用基质匹配校正法通过抵消因对照品溶液与供试品溶液化学组成差异带来的影响等。然而,在实际的分析过程中,使用SPE 对样品进行净化时步骤繁琐,易在各步骤中引入难以预估的误差;同位素内标稀释法则存在内标价格高昂且获取不易的问题,难以全面匹配所有待测化合物。相比之下,基质匹配校正法操作简便,使用样品基质配制对照品溶液校正基质的抑制或增强作用,能够有效地消除基质效应[25],无需繁琐净化步骤与昂贵的内标,适用于快速分析,目前已被广泛应用于各领域,例如在《中国药典》(2020 年版)四部 通则2341 中,规定了使用基质匹配校正法进行禁用农药的LC-MS/MS残留分析[26],在由国家药品监督管理局发布的《化妆品中本维莫德的测定》(BJH202101)中也采用了基质匹配校正法测定膏霜化妆品中的本维莫德[27]。分别考察了PFASs 在上述7 种化妆品样品中的基质效应,使用公式进行计算:ME=A/B,其中A为基质匹配时化合物的峰面积,B为纯溶剂时化合物的峰面积。采用提取后添加法对7种基质类别空白样品进行基质效应评价,用乙腈和7种空白基质分别配制同一浓度的溶液后上机测定,结果如表2所示。ME 值越接近1,说明基质效应越小,反之亦然。从表2的ME值可以看出,30种待测物在不同化妆品基质中普遍存在基质效应。由于化妆品不同剂型的成分差异较大,如水剂和凝胶化妆品中一般含有小分子醇类作为保湿剂或溶剂[28],其在色谱上的保留较弱,容易与弱保留的PFASs 共流出而造成基质效应;膏霜、乳液、粉状、油状和蜡基化妆品均使用了油脂、蜡及其衍生物作为原料或辅料,起护肤、滋润、赋体等作用[28-29],可在不同的保留时间处流出,造成保留时间相近的化合物不同程度的基质抑制或增强效应。由于化妆品的基质复杂程度高,尽管通过前处理可降低样品基质对仪器的污染,但基质效应仍不可避免,为了有效地消除基质效应对定量结果准确性的影响,实现化妆品中PFASs 的快速且准确地测定,本文采用基质匹配校正法对PFASs进行检测。

表2 30种PFASs在不同化妆品样品中的基质效应Table 2 Matrix effects of 30 PFASs in different cosmetic samples
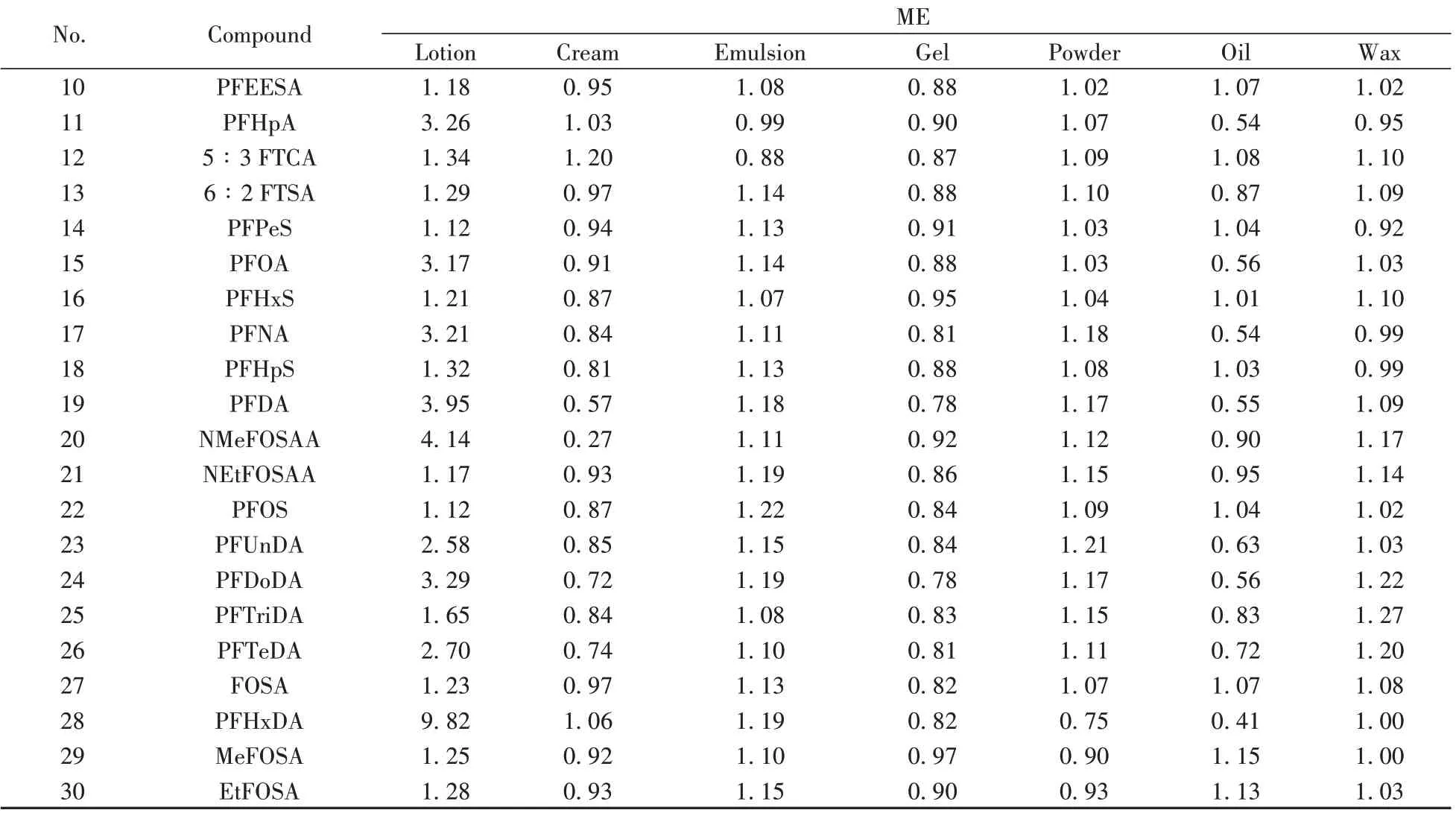
(续表2)
2.5 线性关系、检出限及定量下限
采用本方法测定系列的基质匹配标准溶液,以各PFASs 的峰面积(y)对其质量浓度(x)进行线性回归。结果表明,30 种PFASs 在相应的范围内线性关系良好,相关系数(r2)为0.990 3~0.999 8。分别以信噪比(S/N)≥3 和S/N≥10 倍时的浓度作为检出限(LOD)和定量下限(LOQ),本方法中30 种PFASs 在取样量为0.2 g 时的LOD 为0.025~0.125 μg/g,LOQ 为0.050~0.250 μg/g(见表3),可满足日常的检测要求。

表3 30种PFASs的线性关系、检出限与定量下限Table 3 Linear relationships,LODs and LOQs for 30 PFASs
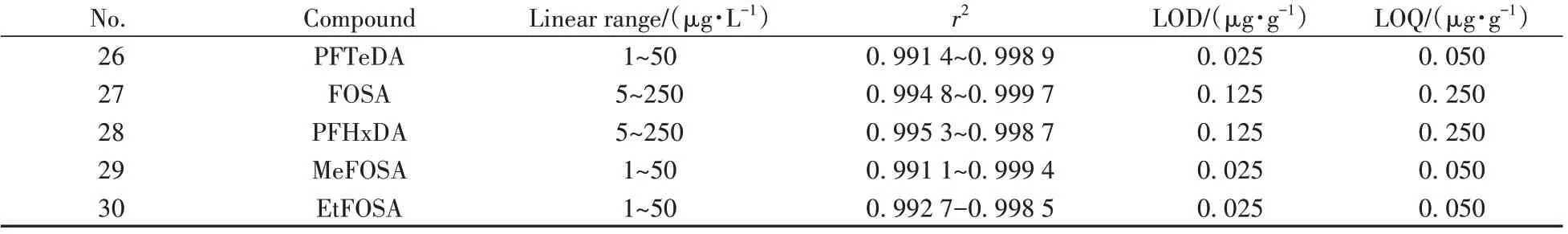
(续表3)
2.6 准确性与重复性
为了考察方法的准确性和重复性,在空白样品中添加3 个不同水平(0.10、0.25、1.00 μg/g)的标准溶液,按照“1.3”方法进行处理,每个加标水平测定6次,计算各化合物的加标回收率考察方法准确性,以含量测定值的相对标准偏差(RSD)考察方法重复性。由表4 可知,在3 个加标水平下,30 种PFASs的回收率为70.8%~112%,RSD 为0.50%~12%,表明该方法能够满足方法学中对准确性和重复性的要求。
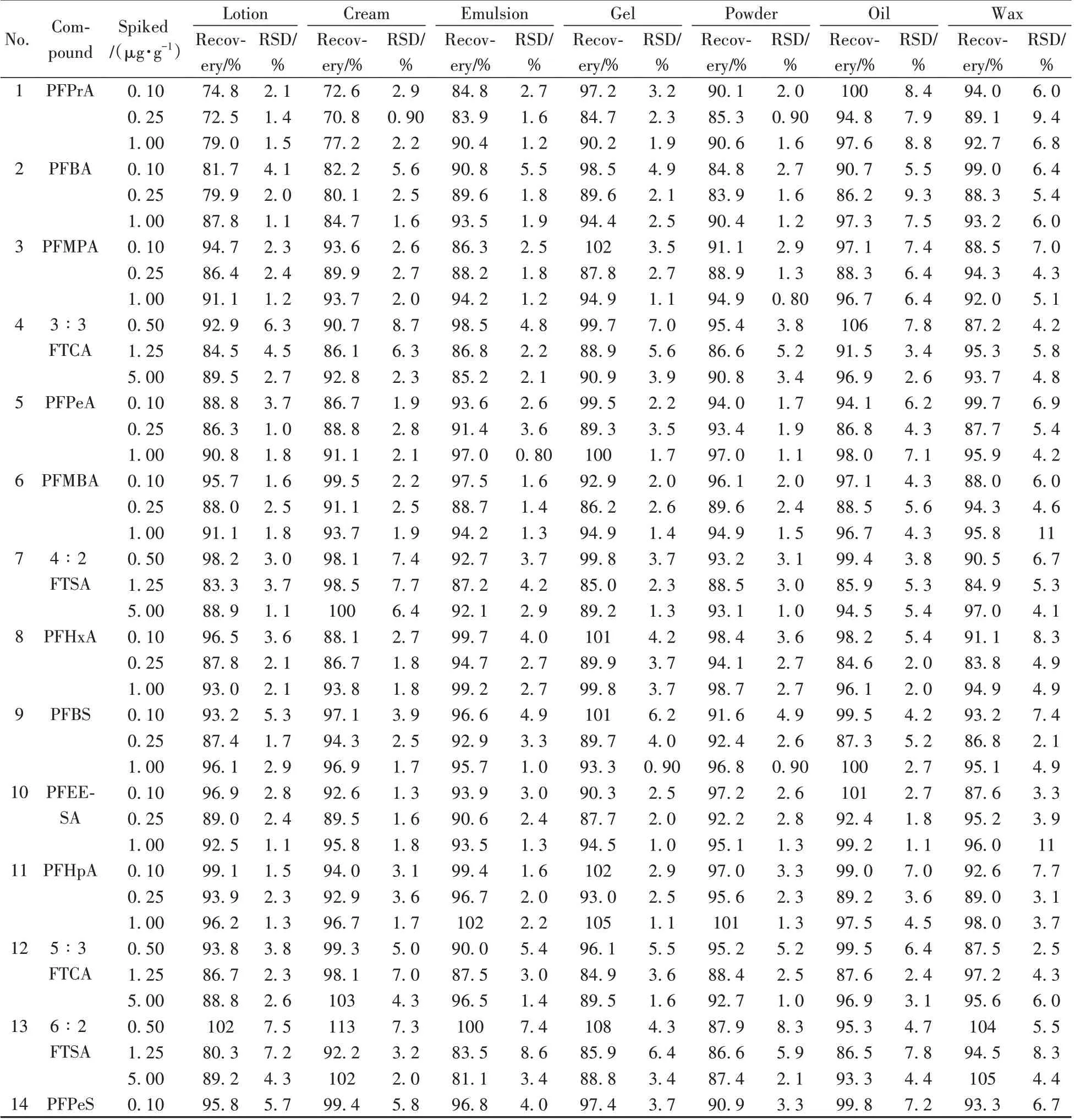
表4 PFASs的加标回收率与相对标准偏差(n=6)Table 4 Recoveries and RSDs of PFASs(n=6)

(续表4)
2.7 方法稳定性
取同一浓度不同空白基质配制的对照品溶液,分别于0、5、10、15、20、24 h 进样,以各PFASs峰面积的RSD(n=6)考察待测物在各类基质中的稳定性。结果显示,在24 h内各PFASs峰面积的RSD均小于10%,表明本文所建立的方法具有较好的稳定性。
2.8 实际样品测定
采用本方法对国内市售的88批次化妆品样品进行检测,其中包含水剂样品10个,膏霜样品14个,乳液样品15 个,凝胶样品7 个,粉剂样品11 个,油状样品15 个,蜡基样品16 个。通过与对照品溶液中各化合物的保留时间、定量离子对、定性离子对及其比值比对进行定性判定,发现1 个阳性乳液样品,检出的PFOA 和PFOS 含量分别为4.7 μg/g 和1.4 μg/g,阳性样品的典型总离子流图如图3所示。PFOA 和PFOS 为两种典型的PFASs,因其强表面活性和良好的疏水性而被广泛应用于包括化妆品在内的各工业领域中[30]。鉴于PFASs 不容忽视的毒性作用,为保证化妆品的安全质量,应重视化妆品中PFASs的控制。
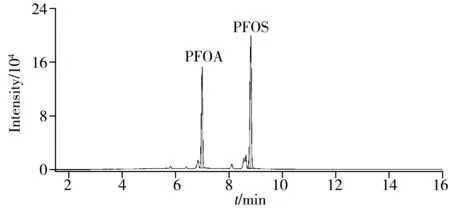
图3 1阳性样品的总离子流图Fig.3 Total ion chromatogram of a positive sample
3 结 论
本研究通过优化仪器条件与前处理条件,建立了超高效液相色谱-串联质谱法测定多种类型化妆品中PFASs的分析方法。该方法具有前处理方便快捷、灵敏度高、精密度与准确度好的优点,能够满足化妆品中PFASs的快速、准确测定要求,为化妆品的安全质量监控和日常监管提供了技术支持。
猜你喜欢
中国环境科学(2023年9期)2023-09-23 12:10:20
食品安全导刊(2020年21期)2020-09-07 09:14:04
农家科技中旬版(2019年9期)2019-10-08 05:27:47
山西农业科学(2019年6期)2019-06-19 07:14:40
中国蜂业(2018年4期)2018-05-09 06:25:08
山东工业技术(2016年13期)2016-06-29 09:05:13
当代化工研究(2016年6期)2016-03-20 16:21:46
化工生产与技术(2016年5期)2016-03-13 10:07:26
化工生产与技术(2016年5期)2016-03-13 10:07:26
化工环保(2014年6期)2014-04-04 10:53:28
