
Potocki-Lupski 综合征1例报道并文献复习
2023-11-07 01:39:18吴剑云钟秋兰吴玉娇朱月娥张敏仪敖丽云
岭南急诊医学杂志 2023年5期
吴剑云 钟秋兰 吴玉娇 朱月娥 张敏仪 敖丽云
PTLS 为一罕见病,发病率约为25,000 分之一,该综合征的特点是婴儿低张力、喂养不良、认知障碍、心血管异常、语言障碍和行为障碍,遗传方式为常染色体显性遗传,大多数受试者为新发生的复制[1]。PTLS 由染色体17p11.2的微重复引起的,通常长度为3.7 Mb。具有广泛的临床表达性,没有特异性的临床表现[2]。由于本病缺乏特异性表现,早期临床确诊比较困难。本文回顾性分析1 例经分子遗传学确诊的PTLS 患儿的临床资料,并复习相关文献,以期提高对此病的认识。
1 病例资料
1.1 病史及检查患儿男,11+个月,系G3P2,孕39+4 周剖宫产,出生体重3.15 kg,身长50 cm,头围34 cm,右手拇指多指,左手拇指并指畸形,生后有喂养困难史。2-个月时每次吃奶30 ml-60 ml,每次吃奶时间30-60 分钟,日总奶量250 ml-300 ml,肌张力弱,反应稍差,睡眠时间多;2+个月抬头能力差;3 个月大时只有4.0 kg,在7+个月大的时候,还不能在没有支撑的情况下坐稳;11+个月时会坐、会爬,每次吃奶50 ml-60 ml,偶尔80-100 ml,每次吃奶时间30-60 分钟,日总奶量400-500 ml。父母非近亲结婚,否认家族遗传病史及类似疾病,患儿有一姐姐健康无异常。查体:体重6.2 kg,身长69.8 cm,头围43 cm。三角脸,眼裂窄,眼睛的外角向下,宽额头和小下巴,眼距宽(图1)。心肺腹无明显异常。检查:头颅MR+DTI+MRS、脑电图、甲状腺彩超无异常;超声心动图提示房间隔过隔血流信号,卵圆孔未闭与房间隔缺损相鉴别;听性脑干反应提示左耳感音神经性耳聋。化验:患儿出生3+个月内多次甲状腺功能检查TSH 增高波动于5.62 μIU/mL-9.11 μIU/mL 之间(4 个月后一直服用优甲乐,TSH 正常);血糖曾有一过性降低:出生3 天时2.65 mmol/L,2+个月3.24 mmol/L;遗传代谢病串联质谱筛查未见特殊代谢产物异常升高。患儿3+个月时予罕见病专项全外显子组测序检测,5 个月时予口咽部吞咽困难等康复治疗。
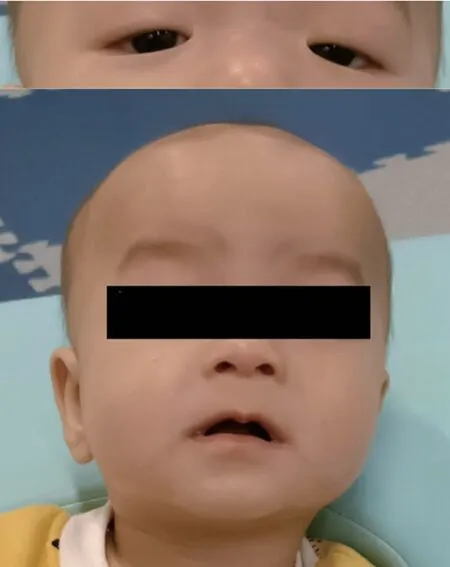
图1
1.2 基因检测(1)检测项目:罕见病专项全外显子组测序检测(三人家系)。(2)检测技术:对受检者基因组DNA进行全外显子组捕获和测序。(3)分析内容:基于二代测序数据进行单核苷酸变异、小片段插入缺失变异和大片段拷贝数变异分析。(4)检测结论:检测到可以解释受检者表型的致病变异。
基因测序数据分析结果提示其基因组DNA 中17P11.2区域存在约3.74Mb 的拷贝数重复变异。该拷贝数重复区域主要包含受检者基因组DNA 上RAI1,FLCN,ALDH3A2等功能基因。该拷贝数重复区域包含17p11.2recurrent(SMS/PLS)region(includes RAI1)(ISCA-37418),为ClinGen 已知的三倍剂量敏感区域(PMID:17357070,20188345等)。该拷贝数重复变异与Potocki-Lupski 综合征(Potocki-Lupski Syndrome;PTLS)[MIM:610883]相关。父母样本经NGS 测序均未见相应拷贝数重复区域的变异,提示该变异可能为新发变异。
2 讨 论
PTLS 是一种非常罕见的染色体异常,由17 号染色体短臂部分重复(17p11.2 微重复)引起。PTLS 首次报道于2007 年,尽管第一例被描述的病例似乎是在1996 年。该病的命名与描述它的两位研究人员Lorraine Potocki 和James R.Lupski 有关[3]。医学文献中报道的PTLS 患者不到100 人[4]。该综合征大多数为散发病例,少数为家族遗传[5]。本例患儿父母样本经NGS 测序均未见相应拷贝数重复区域的变异,提示该变异可能为新发变异。
PTLS 以认知、行为和医学表现为特征。在认知方面,大多数人表现为发育迟缓,后来符合中度智力障碍的标准。在行为上,可能会出现注意力、多动症、退缩和焦虑问题;有些人符合自闭症谱系障碍的标准。医学上,肌张力低下、口咽吞咽困难导致生长迟缓、先天性心脏病、与生长激素缺乏相关的低血糖和轻度畸形面部特征。医学表现可获得婴儿期PTLS 的识别;但对只有行为和认知表现的患儿可能在童年后期才被发现。PTLS 的发育障碍从轻度到重度不等,其临床表现变化很大,特别是在认知水平和行为表型方面[6]。本例患儿特殊面容,肌张力低下、喂养困难导致生长迟缓等表现均符合PTLS 的临床特征,并通过这些早期症状于4+个月时经NGS 测序确诊。该患儿尚小,在认知和行为异常方面,有待进一步观察和评估。
Franciskovich R[4]对37 例PTLS 患者进行 队列性回顾性分析,以调查身材矮小的病因:大约25%(9/37)的PTLS患者身材矮小。其中两名有明确的生长激素缺乏症,另外三名身材矮小且无生长激素缺乏症记录的患者,接受了生长激素治疗,线性生长得到改善。早期识别生长激素缺乏症可以促进患儿的潜在治疗效果,包括肌肉骨骼的线性生长以及低血糖情况下认知的发展。本例患儿亦存在身材矮小,出生2+个月内血糖曾有一过性降低。还需定期监测评估患儿生长发育情况,包括接受生长激素评估或提供生长激素治疗。PTLS 的鉴别诊断包括Smith-Magenis 综合征(SMS)、唐氏综合征、Williams 综合征、短指智力缺陷综合征(del 2q37)、Prader-Willi 综合征或Sotos 综合征。需要排除的第一种情况是SMS,这是一种罕见的综合征,具有类似的临床表现,且涉及17 号染色体(RAI1 基因缺失或突变)。PTLS 和SMS 的发生都是因为一个非等位基因同源重组缺陷,涉及17p11.2 染色体区域的1.3Mb-3.7 Mb,其中发现了RAI1 基因[3]。RAI1 是两种综合征的主要表型特征的剂量敏感基因,PTLS 和SMS 都表现为非特异性发育迟缓、语言障碍和智力残疾等,PTLS 的表现较SMS 轻[7]。国内有研究认为染色体17p11.2 缺失/重复是中国发育迟缓患者中较为常见的基因组疾病[8]。
对于患有PTLS 的患儿没有特定的治疗方法。每个患儿都将受益于适合他需要的个性化治疗,包括语言治疗,物理治疗,行为和交流治疗,或不同的专业教育服务。在这种情况下,遗传建议是必不可少的[4]。对患儿的处理涉及到多学科的照护及今后的常规监测评估[2]。本例患儿通过早期吞咽、喂养和运动等综合康复治疗而得到改善,11+个月时会坐、会爬,现仍在康复治疗中。
综上所述,PTLS 的临床表型无特异,很难进行明确的临床诊断,及时行NGS 全外显子组测序技术以早期明确诊断,有助于及时进行发育干预和医学治疗,最终改善临床结果。
猜你喜欢
心电与循环(2021年4期)2021-11-29 02:41:56
河北医学(2021年10期)2021-10-27 00:37:14
天津医科大学学报(2021年1期)2021-01-26 00:57:22
心电与循环(2020年3期)2020-06-18 13:43:12
中国生殖健康(2020年6期)2020-02-01 06:29:00
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
中国生殖健康(2018年6期)2018-11-06 07:09:38
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
中国医疗美容(2015年4期)2015-04-27 02:24:00
发明与创新(2015年25期)2015-02-27 10:39:16
