
以急性溶血性贫血并急性胰腺炎起病的肝豆状核变性1例病例报道及文献复习
2023-11-07 01:39:18何雪玉舒方华林雪梅陈培德李晓娟廖海霞李栋方
岭南急诊医学杂志 2023年5期
何雪玉 舒方华 林雪梅 陈培德 李晓娟 廖海霞 李栋方,*
肝豆状核变性(hepatolenticular degeneration,HLD),又称Wilson 病(Wilson disease,WD),是一种常染色体隐性遗传性疾病,致病基因ATP7B 定位于13 号染色体长臂(13q14.3)。其特征是铜转运蛋白:三磷酸腺苷酶7B(ATPase copper transporting beta,ATP7B)的功能破坏以及铜的毒性积累而导致的铜代谢障碍性疾病。现就一例以急性溶血性贫血并急性胰腺炎起病的罕见HLD,总结诊治经验,并进行文献复习,本研究已通过中山大学孙逸仙纪念医院深汕中心医院医学伦理委员会审查(2023-SSKY-579)。
1 病例资料
患儿,女,11 岁,因“反复呕吐5 年,加重伴皮肤黄染、排茶色尿3 天”入院。5 年前在当地医院诊断“胃炎”。2年前体检时发现“肝炎”。对症治疗后呕吐症状反复。3个月前呕吐加重伴腹痛。2 个月前查血常规血红蛋白(Hb)156 g/L。3 天前呕吐、腹痛突然加剧伴皮肤黄染、排浓茶色尿,查“总胆红素(TBiL)299.5 μmol/L,Hb 116 g/L”,转至我院,查Hb107 g/L,TBiL 283.1 μmol/L,血淀粉酶1216 U/L,腹部CT 示“脾脏体积稍增大;脂肪肝征象”,予“补液、护胃、抗感染、止呕”等对症后呕吐好转,仍有腹痛。精神、睡眠、食欲差,大便正常,体重下降2.5 kg。查体:生命体征平稳,神疲,反应好,发育正常。皮肤、巩膜黄染,腹软,剑突下、上腹、脐周轻压痛,无反跳痛,Murphy 征(-),肝脾肋下未扪及,余查体无特殊。查:脂肪酶4770.0 U/L, 血淀粉酶1216 U/L,尿淀粉酶8074.4 U/L)ALT、AST、总胆红素均高,贫血加重(Hb 95 g/L,大细胞性贫血),尿潜血阳性,APTT 正常,PT、TT 稍延长,Fbg 低。地贫常规大致正常。初步诊断“1.急性胰腺炎2.贫血、黄疸查因”。
住院检查:血锌卟啉、高敏PNH、血沉、自身免疫性肝病抗体谱、抗核抗体、抗双链DNA-IgG、抗核抗体谱均阴性。病原学、传染病三项、肝炎系列、肾功能指标未见异常,Coombs 实验阴性,G6PD 活性正常、自身免疫性肝病抗体谱等免疫检查无异常,骨髓常规示增生性贫血。上腹部MRCP:考虑肝功能损伤。综合上述,不能排除HLD。进一步查血清铜蓝蛋白:82.1 mg/L(低于正常值),24 小时尿铜定量:1341.96 μg/24 h(高于正常值)。眼科裂隙灯下角膜K-F 环阳性(图1)。头颅MR 未见异常(图2)。肝豆状核变性ATP7B 基因测序提示:ATP7B 基因上变异所致疾病的临床表征与受检者临床表型相符,且变异评级为致病的变异(图3)。且患儿存在两个基因位点的致病变异(图4):其一变异遗传自母亲;另一变异极可能为新发变异,也为致病变异。此外,患儿父亲和哥哥的相应位点未检测到变异基因。
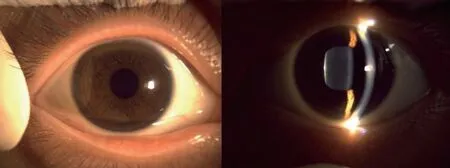
图1 角膜色素环(K-F 环)阳性
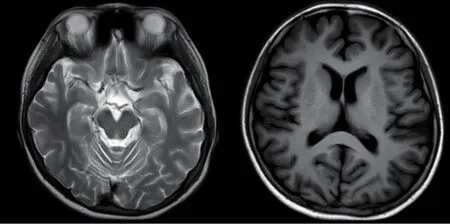
图2 头颅MR 平扫未见异常
患儿接受青霉胺和锌剂驱铜治疗,同时予护肝、抑制胰腺分泌等对症支持治疗后症状消失,住院20 天后贫血明显改善,肝功能、胰腺炎相关指标均恢复正常,无神经系统损害。出院后坚持驱铜和低铜饮食,半年后患儿出现情绪低落超过1 个月,予心理疏导和抗焦虑干预治疗后缓解。

图3 肝豆状核变性ATP7B 基因测序检测结果:检测到可以解释受检者表型的致病变异
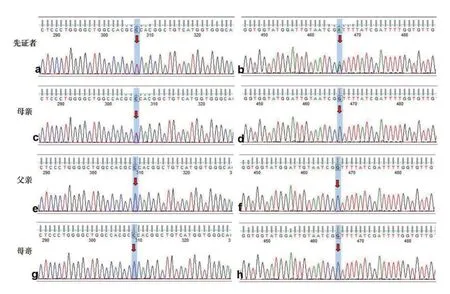
图4 a:ATP7B 基因 染色体位置:chr13:52520505 变异信息:c.2975C>T(p.Pro992Leu)——先证者测序:杂合;b:ATP7B 基因 染色体位置:chr13:52523835 变异信息:c.2828G>A(p.Gly943Asp)——先证者测序:杂合;c:ATP7B 基因 染色体位置:chr13:52520505 变异信息:c.2975C>T(p.Pro992Leu)——母亲测序:杂合;d:ATP7B 基因 染色体位置:chr13:52523835 变异信息:c.2828G>A(p.Gly943Asp)——母亲测序:野生型;e:ATP7B 基因 染色体位置:chr13:52520505 变异信息:c.2975C>T(p.Pro992Leu)——父亲测序:野生型;f:ATP7B 基因 染色体位置:chr13:52523835 变异信息:c.2828G>A(p.Gly943Asp)——父亲测序:野生型;g:ATP7B 基因 染色体位置:chr13:52520505 变异信息:c.2975C>T(p.Pro992Leu)——哥哥测序:野生型;h:ATP7B 基因 染色体位置:chr13:52523835 变异信息:c.2828G>A(p.Gly943Asp)——哥哥测序:野生型。
2 讨 论
HLD 全球患病率约0.25/1 万-4/1 万[1],由于外显率的差异及可能因无症状而未被发现者,实际上患病率可能比临床诊断高3-4 倍[2],任何年龄可发病,以儿童、青少年多见[3]。患者临床表型多样,40%-70% 患者初始表现涉及肝脏[4],且女性较为多见。因受累器官和程度不同而症状各异,可以引起心、肝、肾、关节等器官损害,也可以引起女性闭经、流产,较少见的表现可有甲状旁腺功能减退、胰腺炎等内分泌异常等。即使是同一系统发病者,HLD 仍可出现多种多样的症状。因此,HLD 急性发作的诊断困难,误诊率高[5]。首发症状多种多样[6],但目前国内外文献未见同时以溶血性贫血和胰腺炎起病的HLD 报道。
在胡纪源等[7]的研究中,59.8%HLD 入院前被长期误诊。HLD 可根据肝脏和神经系统症状、体征和实验室检查结果帮助诊断,包括血清铜蓝蛋白、24 h 尿铜、血清铜检测,特别是角膜K-F 环阳性。本例患儿长期被误断为“胃炎”,既往的肝炎病史亦极有可能是急性溶血性贫血发作,可见帮助基层医生对该病早识别、早期诊断和治疗对其预后非常重要[8]。伴有急性血管内Coombs 阴性溶血性贫血可以是儿童HLD 引起溶血的典型特征还可能是首发症状。
溶血可急性发作,也可呈阵发性或慢性病程。其可能机制是铜的促氧化作用[9]。急性溶血(HA)与肝细胞坏死将铜释放到血液中,过多的铜离子损伤红细胞膜而发生氧化损伤、红细胞代谢改变,以及坏死肝细胞释放铜的毒性作用导致抗氧化状态严重受损的结果[10]。HLD 引起的急性肝衰竭(ALF)常合并溶血性贫血溶血[1]。因此,对于病因不明的溶血性贫血并伴有肝脏肿大、肝功能异常、脾功能亢进的患儿,需考虑HLD 可能。类似本案例患者中以急性溶血为主要表现的HLD 误诊率极高,合并急性胰腺炎更是罕见。
急性胰腺炎(AP)在临床急症多见,最常见的诱因是胆道疾病、高脂血症、饮酒,而自身免疫性、血管炎性、药物性、肿瘤、感染、代谢因素、创伤性、医源性因素等也可导致急性胰腺炎。临床上可见溶血性贫血合并有胰腺炎的报道,多认为是溶血性贫血诱发胰腺炎的发生,但国外也有个别病例认为溶血性贫血是胰腺炎的并发症[11]。本病例患儿出现的急性溶血性贫血、急性胰腺炎与肝豆状核变性发病之间的因果关系仍值得进一步探讨。另外,在临床随访过程中,我们除要关注HLD 并发症和药物副作用外,应关注青少年患儿的精神心理状态,开展多学科合作诊治有利于该疾病良好的预后结局。
猜你喜欢
现代临床医学(2021年4期)2021-07-31 07:56:08
昆明医科大学学报(2021年5期)2021-07-22 07:32:50
科学之谜(2019年3期)2019-03-28 10:29:44
中兽医学杂志(2019年1期)2019-01-06 23:48:45
科学之谜(2018年8期)2018-09-29 11:06:46
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
罕少疾病杂志(2016年4期)2016-03-11 16:34:38
首都医科大学学报(2015年4期)2015-12-16 13:00:08
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
药学与临床研究(2015年4期)2015-06-05 11:35:52
