
萝卜硫素干扰微管与线粒体动力学抗肿瘤机制
2023-06-05 07:32周妍吴巍,2*
首都医科大学学报 2023年3期
周 妍 吴 巍,2*
(1.首都医科大学基础医学院生物化学与分子生物学系,北京 100069;2. 肿瘤侵袭和转移机制研究北京市重点实验室,北京 100069)
肿瘤的异质性给肿瘤化学药物治疗(以下简称化疗)带来了障碍,没有一种抗癌药是万能的,针对不同的肿瘤和不同的患者用不同的药物或采取联合用药策略是必要的[1]。因此,研发和筛选更多高效低毒的化疗药物,便于精准地进行个体化疗法,提高治疗效果。现在很多的化疗药物都是针对性地打靶一些关键分子,破坏肿瘤细胞内蛋白网络系统和蛋白稳态,因此发现必要的肿瘤形成机制,探索抗癌药物的抑癌机制,找到对药物敏感的靶蛋白是首要任务。萝卜硫素及其代谢物(sulforaphanes, SFNs)是低毒、高效的癌细胞凋亡诱导剂。笔者课题组多年来在亚细胞水平对SFNs的抑癌机制进行了一系列研究[2-8]。笔者发现这些食物来源的活性成分有独特的分子结构,能够破坏微管,促发抑癌信号,诸如诱导自噬体形成,抑制自噬溶酶体形成,导致线粒体受损蛋白累积、细胞代谢障碍等,最终引起凋亡[2,5-6]。找到了打靶癌细胞的关键靶点,反过来也能设计更多新的抗癌物质,便于使用类癌芯片优中选优,提高治疗精准度。探索SFNs的亚细胞机制也有潜在的应用价值和经济价值。近年笔者团队已经取得了一些有意义的进展,研究结果发表在CancerLetters、CellDeathDisease及CellDeathDiscovery等期刊上。凭一孔之见,现将一些重要的发现综述如下。
1 SFNs解聚微管触发微管动力学异常
用SFNs处理前列腺癌、人脑胶质母细胞瘤、人非小细胞肺癌(non-small cell lung cancer,NSCLC)等在荧光显微镜下可见微管蛋白呈“鸟巢状”分布[4,6,8]。通过透射电子显微镜能看到细胞内空泡化、核包膜消失、核凝集、核碎裂和凋亡小体等。分子生物学分析显示,SFNs介导ERK1/2持续磷酸化,进一步活化26S蛋白酶体和caspase3,最终导致多种蛋白质的表达下调、剪切和降解[3,8]。用SFNs处理人NSCLC细胞,分离细胞总蛋白和线粒体蛋白进行高效液相色谱-串联质谱(high-performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)分析,结果显示在全细胞中有200个蛋白表达下调;在线粒体中有31个蛋白表达下调。使用Uniprot数据库通过生物信息学聚类分析,发现这些差异蛋白中细胞骨架蛋白、线粒体跨膜转位蛋白及线粒体蛋白水解酶等与NSCLC的恶性程度及转归等有关[9]。免疫共沉淀和免疫荧光染色均证明SFNs介导ERK1/2磷酸化,下调α-微管蛋白(α-tubulin)。这种下调源于26S蛋白酶体的活化;caspase3体外剪切试验证明,α-tubulin也能被caspase3剪切[7-8]。SFNs介导的α-tubulin下调加剧了微管的解聚。微管是由α-tubulin和β-tubulin聚合构成的管状结构。在正常生理条件下,微管的聚合和解聚处于动态平衡[10]。一些微管抑制剂可打破微管动力学平衡,诱导细胞凋亡[11]。有报道[12-13]显示,紫杉醇可与微管内表面βⅢ-tubulin结合,促进微管动力学失衡,抑制细胞生长,诱发凋亡。SFNs分子中异硫氰酸基团也可与α-tubulin中的半胱氨酸残基共价结合,引起α-tubulin结构改变,诱发NSCLC细胞凋亡[14]。组织芯片[6]结果显示:正常肺组织和癌旁组织中α-tubulin表达较低,肺癌组织中α-tubulin表达显著升高,且这种升高与肿瘤恶性程度呈正相关。α-tubulin被认为是癌症分级和预后的生物标志物。总之,SFNs介导α-tubulin降解,导致微管断裂,癌细胞凋亡。作为微管的主要成分,α-tubulin在细胞运动、有丝分裂、细胞内运输、细胞骨架重塑和细胞周期中发挥重要作用。SFNs还能降低胶质母细胞瘤G0/G1期CDK4/CDK6的表达,降低G2/M期α-tubulin的表达[3]。通过分析TCGA和CGGA数据库,胶质瘤中CDK4/CDK6和α-tubulin的mRNA表达显著高于正常组织,并与病理分级和临床预后显著相关[3]。蛋白印迹分析显示,在U87MG和U373MG人脑胶质母细胞瘤细胞中,SFNs下调α-tubulin,导致微管破坏和聚集,同时CDK4/CDK6与G1/S-特异性周期蛋白-D1(Cyclin D1)的共定位显著降低[3]。此外,SFNs下调CDK4/CDK6和p-Rb,这种下调被ERK1/2阻断剂PD98059逆转,表明这种下调由活化的ERK1/2介导。蛋白酶体抑制剂MG132逆转CDK4/CDK6和p-Rb的减少;类似地,SFNs对26S蛋白酶体的上调被PD98059逆转[3]。这些研究表明,SFNs通过磷酸化ERK1/2增加了蛋白酶体降解功能,降低了CDK4/CDK6和p-Rb表达,导致细胞周期停滞。笔者还发现PD98059逆转了SFNs诱导的细胞凋亡,表明SFNs通过活化ERK1/2调节细胞周期相关蛋白的表达诱导细胞凋亡。
微管解聚加剧微管动力学不稳定性,抑制微管聚合。与α-tubulin类似,微管不稳定蛋白stathmin1(oncoprotein18)N末端有多个磷酸化位点,C末端能与α-tubulin/β-tubulin二聚体结合形成三聚体复合物,抑制微管聚合,是另一个潜在的抗癌靶点[15]。stathmin1表达也与恶性肿瘤分级和不良预后相关,磷酸化ERK1/2促使stathmin1磷酸化增加,细胞总stathmin1降低, 加剧微管解聚,促进细胞凋亡[16]。
许多细胞质蛋白都能和α-tubulin相互作用,这可能是因为微管担负运输蛋白质的使命。热休克蛋白70(heat shock protein 70, Hsp70)也能和α-tubulin发生作用,调节细胞内蛋白质功能[6]。经蛋白酶剪切受损的蛋白质能被分子伴侣蛋白Hsp70重新折叠、运输、加工以维持细胞的活性。Hsp70在恶性肿瘤中高度表达与肿瘤发生和演进有关。过度表达Hsp70可维持蛋白质稳态,防止化学药物引起的细胞死亡[17]。通过siRNA敲减Hsp70可增加细胞对药物的敏感性。免疫荧光染色显示,SFNs磷酸化ERK1/2上调Hsp70[6,18]。共聚焦显微镜和免疫沉淀分析[6]表明,SFNs促进Hsp70与α-tubulin共定位和相互作用;敲减Hsp70可增强SFNs诱导的微管断裂,降低LC3 Ⅱ/Ⅰ,促进细胞凋亡。组织芯片[6]分析也显示,α-tubulin或Hsp70表达增加与NSCLC恶性分级相关,表明α-tubulin和Hsp70同为SFNs识别的两个关键靶点,Hsp70可能与α-tubulin协作调节细胞稳态。LC3Ⅱ是另一种微管相关蛋白,是公认的自噬标志物。微管聚合促进细胞器或蛋白质沿着微管轨道运输,微管相关蛋白LC3Ⅱ结合自噬体沿着微管轨道移动,与溶酶体融合形成自噬溶酶体[19]。自噬有利于清除细胞内受损蛋白质,维持细胞内蛋白质稳态。笔者用SFNs处理细胞,α-tubulin、β-tubulin、LC3Ⅱ和Hsp70都能易位到线粒体上。蛋白前体可与线粒体外膜(outer mitochondrial membrane,TOM)和内膜(inner mitochondrial membrane,TIM)上的跨膜转位蛋白形成复合体,即TOM复合物和TIM复合物,将蛋白前体转位到线粒体基质内进行折叠[20]。TOM复合物家族成员之一——外膜转位蛋白TOMM20将蛋白质转运给内膜TIM复合物。TIM复合物的核心亚基TIMM23和TIMM17A可与线粒体内源Hsp70(mitochondrial Hsp70,mtHsp70)结合,使被转位的蛋白质进入线粒体基质[21]。在酵母细胞中,TIMM23通过其C端结构域锚着在内膜上,而其N端则延伸至外膜表面,发挥蛋白受体作用[22]。因此,TIMM23和TIMM17A接受TOMM20转位来的蛋白,将被转位的蛋白交给线粒体基质受体mtHsp70, 完成了从微管到线粒体的蛋白质运输过程[22]。因此,细胞凋亡与线粒体蛋白输入有着必然联系。
2 SFNs破坏线粒体动力学导致线粒体蛋白稳态失衡
线粒体大约提供细胞能量的80%,维持线粒体蛋白稳定才能保证细胞正常代谢。细胞质蛋白前体和受损蛋白被微管运输到线粒体加工成成熟的蛋白质或被降解清除[23]。除了经泛素蛋白酶体降解,线粒体蛋白酶水解和线粒体自噬溶酶体清除不能折叠的蛋白是维持线粒体蛋白稳态的重要途径[24]。线粒体离子肽酶 1(lon peptidase 1,LONP1)是线粒体基质中的主要蛋白酶之一,是线粒体看门蛋白(gatekeeper)[25]。在正常细胞中LONP1能够与线粒体跨膜转位蛋白及被输入的细胞质蛋白形成转位复合体,负责将未折叠或受损的蛋白质导入线粒体基质, 并且还能利用自身的蛋白水解酶活性降解不能折叠的蛋白,保证线粒体蛋白稳态[25]。线粒体基质中含有近500种多肽,几乎都是从细胞质转入线粒体。三种保守的蛋白酶LONP1、ClpXP和m-AAA负责折叠或降解这些输入的线粒体基质蛋白[25]。LONP1可结合从细胞质转入的线粒体蛋白,增加其可溶性,抑制基质蛋白集聚[26]。在肿瘤细胞中,LONP1也可以降解异常或损伤的蛋白,维持细胞线粒体蛋白的稳定。LONP1在多种肿瘤细胞,例如在NSCLC细胞中高表达[27]。LONP1 过度表达有利于线粒体代谢,促进NSCLC细胞增殖。使用LONP1抑制剂或通过敲减LONP1可导致线粒体基质蛋白集聚,细胞氧化磷酸化活性降低,促进凋亡[28]。笔者发现SFNs介导了LONP1下调以及线粒体蛋白水解酶caspase3活化。报道[29]显示,LONP1降低可导致caspase3活化,结果产生大量受损的蛋白质和活性氧(reactive oxygen species,ROS)导致细胞凋亡。这说明SFNs可以通过调节线粒体蛋白酶,破坏蛋白质稳态,导致细胞凋亡。
再次,在民间,成立于2015年的Copykiller公司作为“韩国研究财团”指定的学术研究伦理道德教育机构,也是开展高校教师学术道德教育的一支重要力量,每年承担着大量学术道德教育课程,发挥着不可代替的作用。此外,韩国的“学术道德信息中心”“国家生命伦理政策研究院生命科学研究伦理书斋”以及“高等院校教育协议会”等政府外围机构也承担着部分高校教师学术道德教育工作。
经由我院伦理委员会批准从本院2010年1月至2017年12月接受的终末期糖尿病肾病血液透析患者中,抽取52名,随机将其分为对照组与观察组,均26例。对照组中,男14例,女12例,年龄48~75岁,平均年龄(61.5±13.5)岁,病程2~5年,平均病程(3.5±1.5)年。观察组中,男14例,女12例,年龄47~75岁,平均年龄(61±14)岁,病程2.5~4.5年,平均病程(3.5±1.5)年;两组一般资料比较结果p>0.05,可作比较。
3 SFNs诱导自噬体累积导致线粒体代谢障碍和线粒体损伤
进一步研究[35]显示,线粒体上LC3Ⅱ 可能通过结合溶酶体相关膜蛋白1(lysosome-associated membrane protein 1,LAMP1)形成线粒体自噬溶酶体。SFNs与溶酶体抑制剂巴佛洛霉素A1联合降低LAMP1与LC3Ⅱ的共定位,阻断自噬体和线粒体溶酶体融合,导致线粒体自噬体积累。最终,SFNs引发微管功能障碍,诱导自噬体累积,线粒体蛋白质稳态失衡,细胞凋亡。此外,免疫共沉淀显示,α-tubulin可能与Hsp70、FASN、ACACA、ACLY和6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶4(6-phosphofructokinase-2/fructose-2,6-bisphosphatase 4,PFKFB4)等蛋白相互作用。在线粒体中未检测到26S蛋白酶体,但caspase3被证实能切割线粒体结合的α-tubulin。SFNs激活线粒体caspase3,剪切α-tubulin,降解α-tubulin/β-tubulin二聚体。组织芯片分析[6]显示,α-tubulin可增加促进癌症恶性表型。因此,α-tubulin的降解或剪切可能导致细胞骨架解体、增加凋亡小体的形成,抑制细胞周期的进展和细胞运动。在亚细胞水平,SFNs诱导线粒体蛋白质损伤和抑制线粒体自噬溶酶体形成可能导致线粒体肿胀。因此,微管动力学的不平衡可能干扰从细胞质到线粒体的信号传递。胞质Hsp70可对SFNs刺激作出快速反应,以保持蛋白质构象稳定或运输错误折叠的蛋白质进入线粒体进行降解。笔者还发现,SFNs促进α-tubulin与LC3Ⅱ的结合,同时降低α-tubulin与TOMM20、TIMM23和TIMM17A线粒体膜转运蛋白的相互作用。因此,SFNs抑制自噬体溶酶体的形成可以降低错误折叠的蛋白在线粒体的降解,从而导致自噬体中受损的蛋白累积。线粒体膜中的磷脂主要是心磷脂,位于线粒体内膜上,当线粒体受损时转移到外膜,触发线粒体自噬。笔者发现,用线粒体自噬诱导剂羰基氰化物间氯苯腙(carbonyl cyanide 3-chlorophenylhydrazone,CCCP)处理后,总游离脂肪酸(fatty acid,FA)和线粒体磷脂反向增加[2]。这提示SFNs可降低线粒体中的磷脂水平,从而破坏线粒体膜。NIX和BNIP3都位于线粒体外膜上,这两种蛋白质直接与自噬体膜蛋白LC3Ⅱ相互作用。NIX招募LC3Ⅱ至受损线粒体。SFNs下调NIX和BNIP3,上调线粒体相关的LC3Ⅱ、并抑制NIX与LC3Ⅱ的相互作用,表明SFNs可能抑制自噬溶酶体的形成[2]。因此,SFNs可能通过抑制FA生产和微管介导的线粒体自噬体形成导致线粒体损伤,细胞凋亡(图1)。
微管对形成成熟自噬体至关重要。α-tubulin和β-tubulin被证明是线粒体膜的固有成分,是自噬体与溶酶体融合的必需通道[30]。阻断自噬体与溶酶体融合可导致自噬体降解能力减弱、LC3Ⅱ累积和细胞凋亡。笔者的研究[5]证明,SFNs上调LC3Ⅱ/LC3Ⅰ表达,引起自噬体累积,导致NSCLC细胞凋亡[5]。作为细胞骨架成分,微管负责线粒体的运动、运输和线粒体自噬。微管动力蛋白dynein/dynactin可以调节线粒体裂解蛋白Drp1在线粒体上募集[31];微管抑制剂紫杉醇可以诱导线粒体通透转换孔(mitochondrial permeability transition pore,mPTP)的开放,促进凋亡[32]。在H9c2心肌细胞,微管结构异常可以导致线粒体片段化[32]。同为微管抑制剂,SFNs导致线粒体膜电位降低。用SFNs(15 μmol/L)处理细胞24 h后,线粒体出现肿胀和片段化。LC3Ⅱ能与微管相关蛋白相互作用,并沿着微管运动与溶酶体膜蛋白结合,有利于形成自噬溶酶体[19];微管的损伤阻碍自噬溶酶体的形成,导致线粒体代谢异常、细胞凋亡[33]。线粒体自噬有助于清除受损的线粒体或蛋白以维持线粒体蛋白质稳态。抑制线粒体自噬可以持续产生ROS,导致线粒体损伤[34]。线粒体膜电位的降低是线粒体自噬的信号,去极化的线粒体可稳定线粒体外膜上的PINK1,随后募集Parkin到线粒体,介导蛋白质泛素化降解受损线粒体[34]。反之,PINK1和Parkin蛋白的基因突变可导致多种疾病。损伤蛋白过度累积可导致线粒体膜电位的降低以及线粒体的肿胀和破裂。
SFNs介导自噬体形成,阻断自噬体与溶酶体的融合,导致自噬体累积。这些变化可能涉及多种信号通路。HPLC-MS/MS[2]表明,SFNs调节脂蛋白的表达;高表达脂肪酸合成酶(fatty acid synthase,FASN)与癌症恶性程度和不良预后相关。SFNs通过激活26S蛋白酶体下调FASN、乙酰辅酶A羧化酶α(acetyl coenzyme A carboxylase alpha,ACACA)和ATP柠檬酸裂解酶(ATP-citrate lyase,ACLY)表达;SFN抑制α-tubulin与FASN、ACACA或ACLY的相互作用;SFNs还降低细胞内脂肪酸的产生;敲减FASN可增加线粒体异常和细胞凋亡[2]。此外,SFNs降低线粒体自噬相关蛋白NIX和BNIP3及其相互作用,以及NIX和LC3Ⅱ之间的相互作用。因此,SFNs通过抑制微管介导的脂蛋白活性以及抑制溶酶体与自噬体的融合而导致细胞凋亡[2]。
请在稿件内标注作者真实姓名、通信地址、电话、邮箱等信息,便于及时沟通。来稿请发至biologyteaching@126.com,邮件主题标明“微信投稿”。
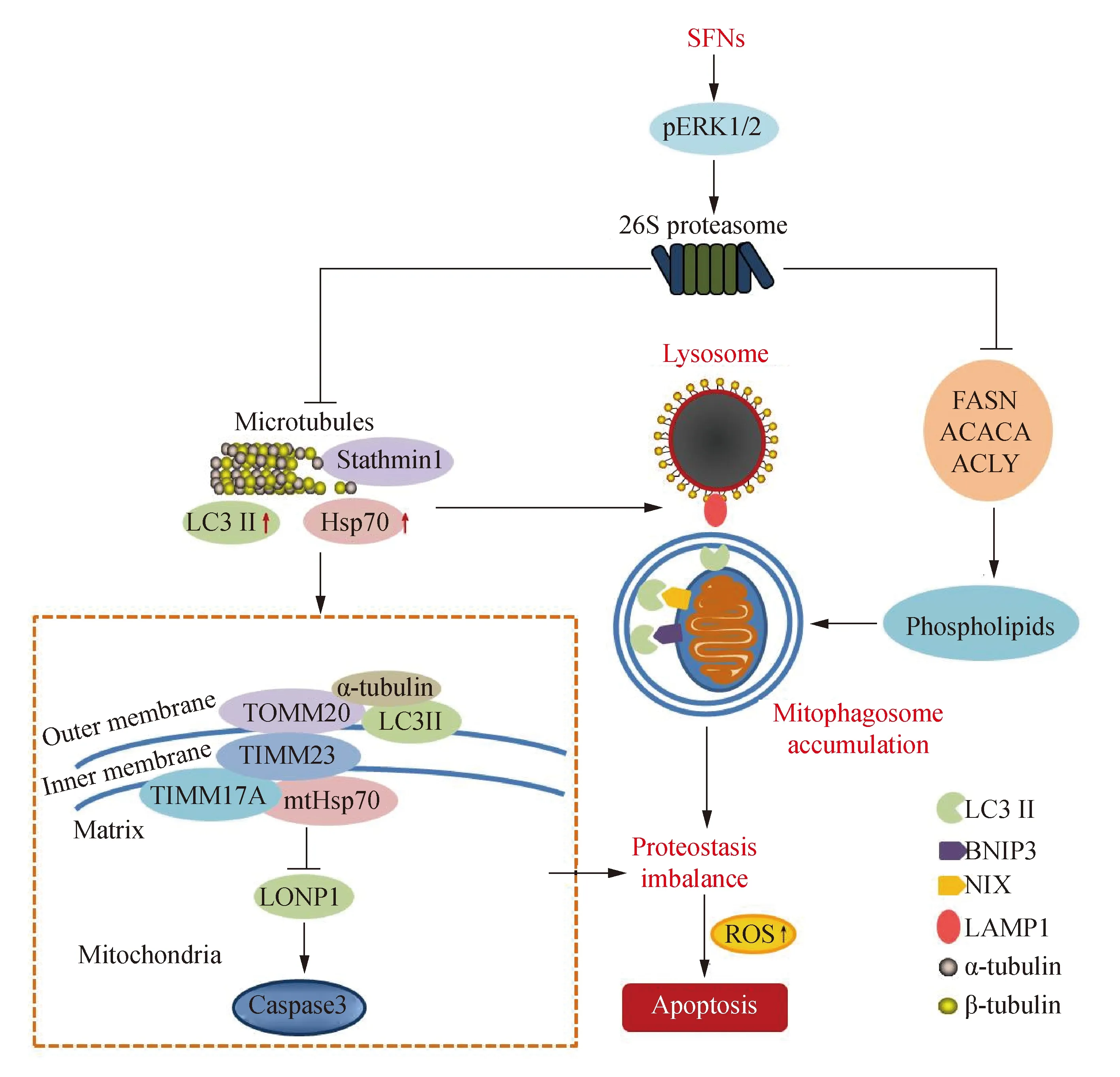
图1 SFNs 干扰微管与线粒体动力学抗肿瘤机制信号转导示意图Fig.1 Signaling map that SFNs cause apoptosis by interfering with microtubules and mitochondrial dynamics in cancer cells
4 SFNs的应用前景和展望
细胞培养对肿瘤细胞的抑制浓度(15 μmol/L)限制了SFNs用于临床试验。然而,笔者的研究[7]证明,SFNs与其他抗癌药物,如紫杉醇联合会将SFNs的有效抑制浓度降低到2 μmol/L,便于临床研究。此外,通过修饰该分子的化学基团或建立新的SFN纳米分子可能会提高工作效率并降低有效浓度。事实上,笔者近期的研究(待发表)显示SFNs在NSCLC和脑胶质瘤细胞中诱导糖酵解酶PFKFB4下调。PFKFB4在许多肿瘤中高度表达,其水平与肿瘤恶性程度有关,因此,SFNs不仅是潜在的候选抗癌药物,而且可能是葡萄糖控制器[36]。SFNs抑制癌细胞信号转导的研究,将帮助我们了解癌细胞运动、生长、自噬及亚细胞代谢等,以便设计更有效的药物来治疗癌症。
利益冲突所有作者均声明不存在利益冲突。
作者贡献声明周妍:整理资料,撰写修改论文;吴巍:提出文章整体思路,撰写论文,总体把关和修订论文。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17
大电机技术(2022年3期)2022-08-06
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29
核科学与工程(2021年4期)2022-01-12
煤气与热力(2021年4期)2021-06-09
生物化工(2021年2期)2021-01-19
生物化工(2020年1期)2020-02-17
中华戏曲(2020年1期)2020-02-12
读与写(2019年35期)2019-11-05
现代职业教育·高职高专(2018年7期)2018-05-14
