
单原子材料的冷冻合成
2022-09-19 06:28:38王茹玥魏呵呵
高等学校化学学报 2022年9期
王茹玥,魏呵呵,黄 凯,伍 晖
(1.北京邮电大学理学院,北京 100876;2.清华大学材料学院,北京 100084;3.华东理工大学化学与分子工程学院,上海 200237)
在催化剂的设计中,提升性能的基础思路主要包括增加有效活性位点和提升本征活性[1,2].单原子催化剂不仅能够实现这两种策略的结合,同时还给出了额外的优势(如同质的活性位点、最大的原子利用效率等[3~5]).因此,单原子催化剂在催化领域占据重要而特殊的地位.单原子催化剂是指孤立的单个原子分散在载体上的催化剂,每个单独的金属原子之间没有任何形式的相互作用,但孤立金属原子和载体上的原子形成了独特的配位结构[6~8].单原子催化剂的活性位点通常由单个金属原子以及载体表面的直接相邻原子或其它官能团协同组成,因此,各个活性位点的催化性能可以相似,也可能不同,这取决于金属单原子与其相邻原子间的相互作用[8~12].在热催化、电催化和光催化等能源转化领域内,单原子催化剂都表现出显著优于传统纳米催化剂的活性、选择性与稳定性[13~16].
研究人员以典型反应为例,从单原子催化剂独特的局域结构与电子性质等角度出发深入探讨了其优异性能的根本原因[17~21].研究表明,单原子催化剂的高反应活性由诸多因素共同决定.首先,与纳米催化剂相比,单原子催化剂的金属原子的不饱和配位键更多,更有利于反应底物的吸附与活化[22~25].其次,单原子催化剂具有独特的量子尺寸效应,其中电子的约束会导致能级增加和前线轨道隙增大,这种优化的金属单原子的电子结构有利于调节活性位点的内在活性[26~32].最后,由于单原子催化剂金属组分的表面自由能增大,金属位点与载体之间的化学作用增强,这使得电荷转移更易进行[33~38].总之,由于低配位状态、量子尺寸效应和原子与载体之间的强相互作用,单原子催化剂不仅具有最大的原子利用效率,而且表现出较高的活性和选择性,并提供了一个简单而有效的模型来探索催化性质,这有助于在原子级别准确合理地设计电催化活性位点,并指导设计单原子催化剂的合成[39,40].
在单原子催化剂的合成过程中,主要面临的困难源于其尺寸效应所带来的高表面能和不饱和的配位环境,导致单原子金属往往处于亚稳态,并伴随易于团聚形成团簇或者纳米粒子的趋势[41~43].科研人员提出了“自上而下”和“自下而上”两种基本合成路径[4,44~48].在“自下而上”的合成路径中,首先选择合适的金属化合物作为前驱体,然后采取措施得到金属单原子,并对其进行限制和捕获,达到锚定作用,防止其迁移和聚集[4,44~48].在该路径中,由于合成方法具有简单和易于大规模生产的特点,湿化学方法成为研究如何制备原子级催化剂的主要选项之一[49~51].
在常规的湿化学合成方法中,最关键的步骤是如何实现前驱体的原子分散和隔离,并将其锚定于载体表面,防止单原子的迁移和团聚.目前主要合成策略包括以下几种:
(1)配位设计策略.利用配位点与金属组分间的强相互作用,合理设计配位原子作为锚定位点以稳定单原子[52~54].如厦门大学郑南峰课题组[55]通过引入乙二醇修饰TiO2纳米片载体,促进对PdCl24−前驱体的吸附和结合,从而通过进一步的光还原制备得到了高度稳定、原子分散的Pd催化剂(Pd1/TiO2).该催化剂在C=C键加氢中表现出高催化活性,其活性超过商业Pd催化剂表面Pd原子的9倍.在此合成策略中,金属原子与载体上配位原子间的强相互作用,不仅有利于提高单原子催化剂的稳定性,还可以通过配位原子对活性中心金属原子的电子结构进行调节,从而提高催化性能.
(2)缺陷工程策略.缺陷的存在会改变周围原子的电子结构和配位环境,导致出现空位和不饱和配位点,因此载体上的缺陷可以作为“陷阱”以捕获金属前驱体和锚定金属原子[56~59].如清华大学李亚栋课题组[60]制备了具有丰富氧空位表面缺陷的二氧化钛纳米片载体.氧空位不仅可以锚定和稳定Au单原子,还促进了独特的Ti-Au-Ti配位结构的形成,该结构可以通过降低能垒和减轻孤立Au原子位点的竞争吸附来提高催化性能.总而言之,缺陷工程应形成均匀的缺陷中心作为锚定位点,从而稳定单原子金属活性中心,因此缺陷工程可以通过控制缺陷浓度来调控金属单原子的负载量.此外,缺陷可以调整活性中心的光学和电子特性,有利于催化剂的协同催化效应,扩大了单原子催化剂的应用场景.
(3)空间限制策略.将单个金属原子空间限制在分子尺度的笼中防止其迁移,也被认为是制备单原子催化剂的有效策略[61~65].此策略一般包括封装金属前驱体与去除配体将其转化为稳定于载体的单个金属原子两步.Gates等[65]选择沸石作为主体,[Pt(NH3)4]2+配合物进入沸石KLTL的孔隙,并优先与K+离子进行离子交换,从而被锚定在沸石骨架上.然后,通过氧化处理获得了定义明确的单原子Pt催化剂,并用于催化CO氧化.该策略的关键在于如何基于沸石、金属有机框架(MOFs)和共价有机框架(COFs)等不同载体,选择具有合适尺寸的分子和电荷状态的单核金属前驱体.
基于上述合成策略,研究人员开发出了包括共沉淀法、浸渍法、强静电吸附法、化学还原法和有机金属配合物法等一系列液相制备单原子催化剂的方法[49~51].然而,这些方法仍存在合成过程复杂、形核机理不清晰、均匀活性位点难以获得及金属载量较低等问题,限制了单原子催化剂的发展和应用.本文综合评述了创新的冷冻合成方法,讨论了一系列的冷冻合成单原子材料的具体实例,介绍了不同合成路径中单原子催化剂的制备机理,并针对其特定催化反应进一步探讨了其高催化性能的来源,从而为高性能单原子催化剂的可控制备提供了借鉴.
1 冰相光还原
在传统的紫外光还原中,通常是直接对金属前驱体溶液进行紫外辐照,此时若不在合成中进行额外的调控,通常得到的是金属纳米晶[66,67].考虑到溶液环境中离子和原子的扩散过程对于金属的形核与生长结晶过程的影响,2017年,本课题组[68]创新性地报道了冰相光还原策略,成功制备了原子级分散的Pt负载型催化剂,开启了冷冻合成的新纪元.具体制备方法如图1所示,首先使用液氮快速冷冻金属前驱体溶液,然后在低温环境下,对冰相的金属前驱体进行紫外辐照.因为冰的晶格可以有效限制反应物离子与产物粒子的扩散,光照结束后,冰块自然融化即可得到分散在溶液中的金属单原子.此外,在该方法中几乎没有金属前驱体的浪费,因此,对于贵金属原子级分散材料的制备具有重要的意义.如图2所示,除了在石墨烯载体上发生了局部团聚,Pt单原子可以在大多数基底上均匀负载且稳定存在.X 射线近边吸收谱(XANES)研究结果表明,在Pt1/MC 中,主要存在Pt—C/O 化学键;在Pt1/TiO2样品中,主要为Pt—O化学键,均未表现出Pt—Pt化学键,由此证实载体表面的Pt金属组分以孤立原子的形式稳定存在,并与球差透射电子显微镜的表征结果相互验证.

Fig.1 Schematic illustration of the iced⁃photochemical process(scale bar:2 nm)[68]
此外,为了探究Pt单原子稳定存在的机理,进一步使用第一原理分子动力学,分别探究了Pt单原子在溶液中和不同基底上的稳定机理.结果表明,Pt单原子在水溶液中会自发地解离水分子,从而形成稳定且不发生团聚的H—Pt—OH 结构[图3(A)];当溶液中引入载体时,H—Pt—OH 结构中的H 和OH 会自发地结合成H2O 并脱去Pt 结构,然后Pt 单原子稳定地锚定于载体的边缘或者缺陷位置上[图3(B)].总之,无论在冰相合成过程的任何环境,Pt均更倾向于形成孤立的原子结构而不是团簇形式.这也是各种原子级分散Pt负载型催化剂制备的基础.

Fig.2 HAADF⁃STEM images of samples[68]

Fig.3 Illustrated structure and partial charge of the Pt single atom in the form of H—Pt—OH in water environment(A),structure and size distribution for Pt single atoms and Pt clusters on mesoporous carbon(B)[68]
所制得的负载在介孔碳上的原子级分散的Pt催化剂,表现出了当时国际领先水平的电催化析氢活性和稳定性.通过密度泛函理论(DFT)计算讨论了催化剂的反应机理,催化剂的高活性可归因于以下几点:(1)与商业Pt/C相比,单原子Pt活性位点的H吸附吉布斯自由能更小;(2)Pt原子被锚定在介孔碳缺陷位点时,二者之间发生电荷转移,Pt带正电荷,展现出未占据的5d态密度,其与H原子的1s轨道会产生强烈的相互作用,这有利于析氢过程中的电子对和氢化物的形成.本课题组[69]还进一步拓展了快速冷冻策略对金属离子输运的调控,并开发了新型的冰系固态电解质.研究发现,金属盐水溶液快速冷却得到的固态冰材料能够传导不同的金属离子(Li+,Na+,Mg2+,Al3+,K+,Mn2+,Fe2+,Co2+,Ni2+,Cu2+,Zn2+,Ba2+).以1 mol/L CuSO4溶液的冰为模型,通过模拟计算,探讨了Cu2+在冰Ⅰh晶体结构下可能的传导路径,并结合交流阻抗谱、直流电沉积和冷冻透射电子显微镜等实验,证明了制备得到的该类固态冰材料的晶格中存在金属离子缺陷,其能够在外部电场的驱动下进行迁移,具有传输离子的能力.该方法不仅为固态电解质的体系进行了补充,还为低温下的电化学器件的设计提供了新的发展思路与广阔的探索空间.
2018年,北京大学郭少军课题组[70]通过类似的冰相光还原策略制备得到了g-C3N4上的高密度Pt单原子,制备原理如图4所示.首先混合了载体与金属前驱体,并通过长时间的搅拌使前驱体吸附在载体上,然后再通过液氮冷冻形成冰块进行后续的光还原过程.由于g-C3N4含有丰富的氮空位,产生了明显的缺电子效应,这显著增强了Pt单原子与载体g-C3N4之间的反应性金属-载体相互作用(RMSI).这种强化学作用有利于稳定金属单原子,以确保高载量与高覆盖密度下Pt仍然以孤立原子状态存在.进一步通过扩展X射线吸收精细结构(EXAFS)表征探究了单原子Pt的配位结构,表明Pt单原子会优先与氮空位位置的二配位碳形成Pt—C 键,这与对载体的理论模型探究结果一致.这种Pt—C 之间的相互作用将使Pt单原子具有更负的5d电子态,加强了Pt单原子与载体之间的RMSI.另外,计算表明在光催化析氢反应中,RMSI形成的Pt—C2C活性位点具有独特的电子与几何效应,因此表现出更强的光生电子捕获能力、更小的H2解吸能及显著优化的析氢能力.
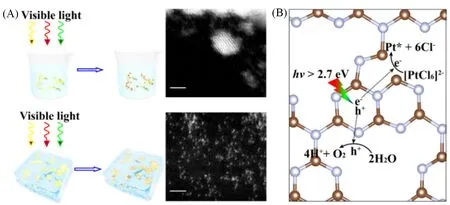
Fig.4 Schematic illustration of the icing⁃assisted in situ photocatalytic reduction method for preparing the supra⁃high⁃density Pt SAs⁃loaded g⁃C3N4[70]
2019年,郭少军课题组[71]继续利用载体表面C2C和与其临近的两个二配位N原子不同的电负性,使其选择性地吸附[PtCl6]2−离子和Ru3+离子,再进一步将溶液冰冻,通过冰相光还原策略制备得到负载在富含N空位的g-C3N4邻近的Pt-Ru单原子催化剂(图5).X射线吸收精细结构(XAFS)研究证实,催化剂中存在邻近Pt⁃Ru 单原子,并且N 空位在邻近Pt⁃Ru 单原子的形成中至关重要.理论模拟也表明,g-C3N4结构中的N空位构成了三角形亚纳米空腔,可通过形成Pt—C和Ru—N键来稳定邻近的Pt⁃Ru单原子.这种C-Pt-Ru-N配位结构不仅具有更负的吸附能和更稳定的化学结构,还通过具备的富电子效应促进了具有还原依赖性的CO催化氧化反应.实验也证实了邻近Pt⁃Ru单原子是催化CO氧化的最优化位点,它可以与O2形成桥型吸附,并通过平衡反应能量具有更高的O2活化性能,最终体现了超高的催化活性.

Fig.5 Schematic illustration of the icing⁃assisted in situ photocatalytic reduction method for preparing PtRu SA⁃CN620(A),HAADF⁃STEM images(B) and the corresponding distribution of C(C),N(D),Ru(E)and Pt(F)of PtRu SA⁃CN620[71]
2021年,斯坦福大学崔屹课题组[72]将IrCl3(H2O)3和Ni9Fe羟基氧化物的混合水溶液在液氮中快速冷冻,并对其进行随后的光还原、离心清洗和冷冻干燥等操作,得到了负载在Ni9Fe羟基氧化物载体上Ir单原子催化剂(图6).DFT计算和XANES结果表明,Ir可能的存在形式包括Ir4+取代Ni4+的传统层内取代掺杂和单个IrO6八面体直接与NiFe层结合两种.DFT理论计算显示高氧化态的Ir位点的加入,提高了OOH*的稳定性,改善了析氧反应机制中Ir位点的第二步(OH*到O*),并导致了NiFe(100)中的Fe位点结合减弱(ΔGOH最正)和OH—OOH尺度的改善,最终导致了水氧化催化性能的提升.

Fig.6 Preparation route to Ir single⁃atom on NiFe oxyhydroxides and atomic structure characterizations of NiFeIr by HAADF⁃STEM[72]
总之,冰相光还原策略为单原子、亚纳米团簇以及团簇等小尺寸材料的可控绿色合成提供了新的途径,也为调整湿化学反应中金属组分的形核与生长开辟了新的视角,提出了在金属单原子与载体间构建RMSI 的新方法,指导了高密度负载型单原子催化剂的制备与电子结构调控,并系统地展示了金属单原子氧化态和活性位点配位环境的可控调谐.该策略有望被拓展到多种传统的液相化学/光化学反应中,并应用于几何结构、电子结构与活性位点精准设计的负载型单原子催化剂的可控生产.
王天放:知乎上有一个问题:人生要怎么做才不后悔?人生怎么过都会后悔。你永远只能走一条,怀念着另一条,但你不能回头。这就是在无知情况下的第二原则:选一条路,一以贯之。
2 冰融化缓释合成
基于经典形核理论,典型溶液合成中均相形核和非均相形核过程中,进入形核状态所必需的活化能(ΔG*)可分别描述为[73~76]
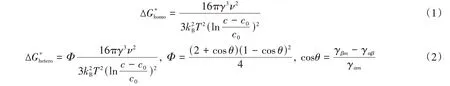
式中:γ(J/m2)为形核的单位表面积的自由能增加;ν(m3)为形核的摩尔体积;kB(J/K)为玻耳兹曼常数;T(K)为反应温度;c(mol/L)为溶液的实际浓度;c0(mol/L)为溶液的饱和浓度;γαm(J),γβm(J)和γαβ(J)分别为结晶相与液体、固体与液体以及结晶相与固体之间的界面能.
由此可知,固态反应产物形核的活化能与溶液的浓度密切相关.这意味着在一定的温度下,降低反应物浓度时,其形核的活化能会增加,形核变得更加困难.故而可以在溶液合成过程中通过降低反应物的浓度来有效抑制液相反应过程中金属固态反应物的形核过程[77,78].
此外,1950年,科学家们已经制定了LaMer 模型来描述均相形核框架,该模型也可以被扩展到解释金属纳米晶的形成[79,80].在图7所示的模型中,一旦金属前驱体被还原或分解,溶液中金属原子的浓度预计随时间的增加而增加,而一旦金属原子的浓度超过形核的最小值(),原子将自发地聚集或组成形成原子核;而金属原子若被快速消耗,低于时,形核行为停止.这个过程是可以动态重复的[80].类似的,2017年,Mirsaidov 等[81]通过原位透射电子显微镜观察发现,金属纳米晶在溶液中合成时分成以下3步:(1)溶液自发分为高浓度相和低浓度相;(2)高浓度相中形核得到非晶的纳米团簇;(3)非晶团簇结晶生长.这进一步证实了反应物的浓度显著影响金属纳米晶的形成.由此可推论通过控制反应物的局域浓度始终处于较低的水平,将有效抑制金属组分的形核与生长.
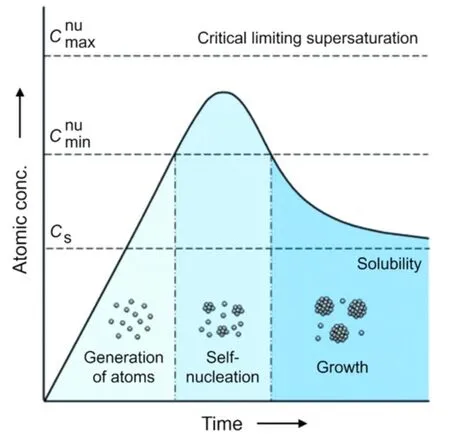
Fig.7 Plot of the concentration of atoms as a function of reaction time illustrating the major steps involved in a synthesis,including the generation of atoms,homogeneous nucleation,and growth[80]
为了实现对溶液中反应过程与形核行为更精准的调控,2018年,本课题组[82]首次提出了冰融化缓释合成策略[图8(A)],并成功制备了多种原子级分散催化剂.区别于传统的液相合成工艺,本策略将冰相反应物A置于液相反应物B中,在0 ℃环境下使A缓慢融化.简化模型计算得到,该方法单位面积单位时间内释放的离子数比传统的化学注射法低了两个数量级.由此可判断,该方法成功地控制了液相环境中的反应物A始终处于低浓度水平,有效减缓了反应动力学与反应产物的形核与生长过程,最终得到原子级分散金属溶液.如图8(B)所示,该策略在制备各种原子级分散的金属材料方面表现出很强的通用性.

Fig.8 Illustration of the syntheses of atomically dispersed metals(A),periodic table of the elements and STEM images for multiple types of atomically dispersed metals,including Co,Ni,Cu,Ru,Rh,Pd,Ag,Os,Ir,Pt,and Au(B)[82]
此外,以金属Ag为例,进一步利用第一性原理分子动力学,计算探究了原子级分散金属的形成机制,如图9(A)~(F)所示.首先,研究发现,Ag原子在低温环境下会被不同的水壳所包围,从而阻止金属单原子向二聚体的转变行为.同时,冰相缓释方法持续提供的低Ag浓度,可以极大程度上减缓金属原子之间因碰撞而导致的团聚现象.总之,Ag原子周围的水壳及其低浓度使原子级分散的Ag溶液可以稳定存在长达约10 h.其次,探究了Ag原子在载体表面稳定存在的机理[图9(G)],计算了Ag单原子与二聚体的相对吸附能量.结果表明,当载体表面存在缺陷(如空位缺陷、扶手状边缘、锯齿状边缘、孔结构等)时,单原子和二聚体的相对能量为负值,即Ag原子更倾向于与在载体表面以孤立原子的形式存在,这与实验结果一致.
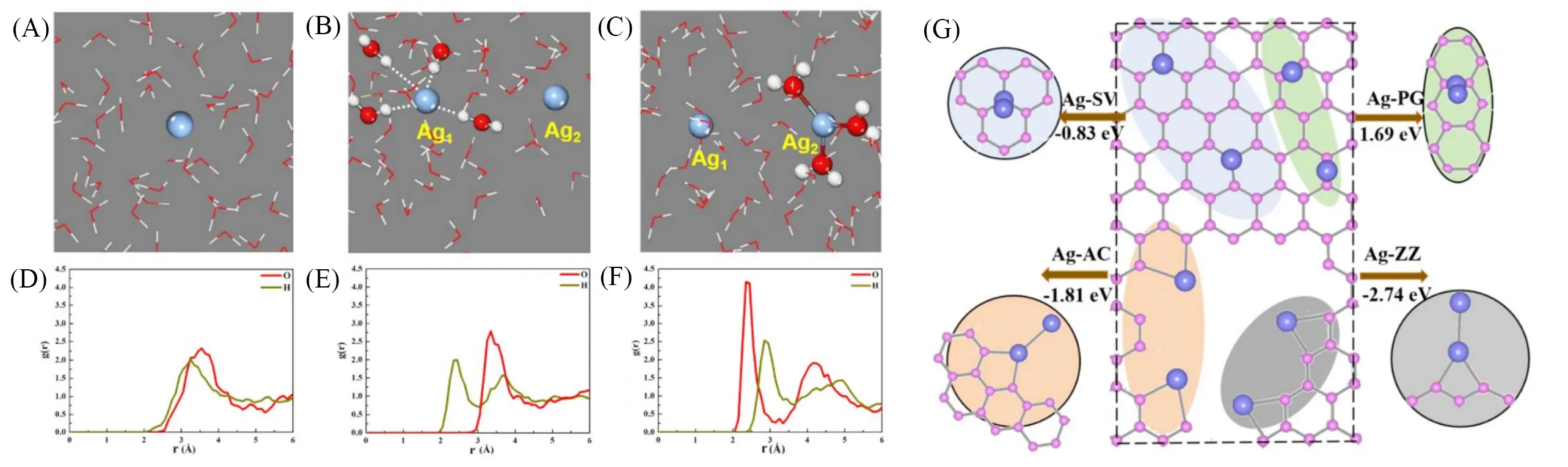
Fig.9 Selected atomic configurations of isolated Ag atoms in an aqueous solution based on first⁃principles molecular dynamics(FPMD)[82]
3 超低温液相合成
由经典形核理论中的式(1)和式(2)可知,在液相合成中形成金属纳米晶所必须跨越的形核势垒不仅与溶液中金属组分的浓度密切相关,还受到反应温度的显著影响[73~76].在保持溶液浓度不变的情况下,进入形核状态所必需跨越的活化能势垒(ΔG*)随温度的降低而增加.推断可知,若在超低温度下进行液相合成,则形核势垒ΔG*显著增加,从而抑制固态反应物的形核与进一步生长,最终得到原子级分散的材料.此外,超低温的反应环境还将影响反应动力学,从而降低形核速率,这有利于金属单原子形态的保持与稳定.
3.1 超低温液相光还原
2019年,本课题组[83]提出了超低温液相光还原策略,具体实施方法如图10所示.对−60 ℃环境下的金属前驱体溶液进行紫外光照还原,超低的反应温度降低了反应动力学,并增大了反应过程中的形核势垒.因此,在这种超低温(−60 ℃)环境的液相反应中,固态反应产物的形核与生长被有效抑制,成功得到了金属单原子溶液,且通过后续的负载、抽滤、清洗以及干燥等流程,最终合成了负载在氮掺杂介孔碳上的金属单原子催化剂.

Fig.10 Schematic illustration of the synthesis of atomically dispersed Pt[83]
通过该策略制备得到的Pt1/NMC催化剂表现出优异的氢析出电催化活性,仅在55 mV的过电势下就实现了100 mA/cm2的电流密度,在40~60 mV 过电势工作范围内,Pt1/NMC 在相同电位下表现出了Pt/NMC团簇催化剂和商业Pt/C 3倍的质量活性.理论研究发现,这是由于原子级分散的Pt具有不饱和配位,这有利于催化性能的提升.此外,Pt1/NMC 单原子催化剂表现出相当出色的稳定性,在0.5 mol/L H2SO4电解液中,经历了5000次循环伏安稳定性测试后,催化剂几乎没有出现析氢性能的衰减和原子级分散Pt的团聚现象.
3.2 超低温液相化学还原
2019年,本课题组[84]同时进行了更为基础普适的超低温液相化学还原合成方法,仅通过简单地将超低温的还原剂溶液滴加至超低温的金属前驱体溶液中,即可通过简单的液相还原反应得到金属单原子溶液(图11).基于此方法制备得到了热稳定的氮掺杂介孔碳负载原子级分散钴催化剂(Co/NMC-LT900).并通过XAFS测量发现,催化剂中的Co原子带正电,并形成了Co-N/C配位结构,且没有明显的Co⁃Co配位,证明液相还原制备得到的原子级分散的Co与NMC基底上的吡啶-N配位,并在热活化后以Co⁃Nx的配位方式被牢牢锚定在氮掺杂介孔碳载体上,并由此具有结构热稳定性.

Fig.11 Schematic illustration of the ultra⁃low temperature solution reduction process[84]
在中性和碱性条件下的ORR电催化领域,单原子催化剂表现出显著超越传统团簇/纳米颗粒催化剂和商业Pt/C的氧还原催化活性和稳定性.通过计算自由能进一步探索了催化剂的氧还原性能,结果表明,Co-Nx活性位点上的ORR过程由4个步骤组成,前3步的热力学均为放热过程,涉及*OH 解吸的最后一步为速率决定步骤,仅需要约0.16 eV非常小的能量,远小于Pt/C催化剂,因此吡啶-N位点吸附的Co单原子位点表现出优越的活性.进一步将催化剂应用于微生物燃料电池器件中,Co/NMC-LT900也表现出了远超文献报道的功率密度[(2550±60)mW/m2]和长达820 h的稳定运行,能够同时实现污水处理与高性能产电.
考虑到载体在锚定单原子方面的显著作用,2019年,本课题组[85]在抑制液相合成中固体产物形核与生长行为的基础上,进一步探究了非均相形核过程,提出了一种在基底上直接锚定原子级分散金属催化剂的一锅法合成策略.但是,由于非均相形核的形核势垒显著低于均相形核过程,实验中必须将反应驱动力保持在较低水平,这给实验带来了严峻的挑战.我们利用−40 ℃的低温液相环境,开发了一步法合成原子级分散催化剂的工艺(图12),并通过尝试多种载体和贵金属,验证了所提出合成策略的普适性.

Fig.12 Schematic illustration of the one⁃pot solution synthesis of atomically dispersed Pt on NMC substrates[85]
本课题组[85]采用分子动力学模拟方法,结合约束极小化技术,针对设想的两个孤立的Pt原子均在乙醇中的形核路径1,与一个Pt 原子在乙醇中、另一个Pt 原子位于NMC 载体上的路径2,分析了低温(−40 ℃)和室温(约25 ℃)下Pt⁃Pt二聚体的非均相形核过程[图13(A)和(B)].计算结果表明,由于低温下原子扩散缓慢,且乙醇分子具有更紧致的结构,与室温下对比,不论反应路径1还是反应路径2,低温下Pt⁃Pt形核过程均需要克服更大的形核势垒.因此,低温环境可以有效抑制Pt⁃Pt二聚体的形核,从而利于形成高度分散的Pt单原子.进一步对Pt单原子在NMC基底上的稳定性进行了理论分析.模型计算表明[图13(C)],原子级分散的Pt与缺陷位点或边缘位置的结合,在能量上比二聚体Pt与载体相同的结合更稳定,说明在NMC载体上Pt也是优先保持孤立原子状态,而不是形成二聚体或团簇.

Fig.13 Schematic illustration of the two types of nucleation pathways of Pt atoms in the ethanol and NMC substrate reaction mixture(A),energy diagram of path⁃2 for Pt⁃Pt dimer formation at -40 ℃(up panel) and RT(bottom panel)(B),schematic diagrams of structures and distributions for atomically dispersed Pt atoms and Pt⁃Pt dimers on sites of NMC without vacancy defects(WVD),single point defects(SPD),double point defects(DPD)and hole vacancy defects(HVD)(C)[85]
制备得到的负载在氮掺杂介孔碳表面的Pt单原子催化剂(Pt/NMC-LT)被证明是一种优异的电化学析氢催化剂.与Pt 团簇(<1 nm)和Pt 纳米颗粒(2~3 nm)商业催化剂相比,Pt/NMC-LT 表现出显著提高的催化活性、反应动力学速率及优异的稳定性.通过DFT系统计算了不同反应构型下H原子在Pt单原子上的吸附自由能,以探究催化剂高效催化析氢反应的机制.与Pt(111)表面对比,Pt单原子催化剂吸附H的自由能更接近最佳吸附自由能,因此,Pt/NMC-LT催化剂具有优异的析氢反应(HER)活性.
2021年,东北石油大学李智君课题组[86]通过低温液相化学还原策略,得到了锚定在有缺陷的六方氮化硼纳米片上的钯原子催化剂[图14(A)~(C)],Pd以Pd1-N3的形式存在.DFT计算确认了空位缺陷可以为Pd原子提供锚定位点,Pd吸附在B空位处时可与周边临近的N原子有更强的结合作用,而Pd4d与N2p在费米能级附近具有明显的轨道杂化现象,也说明了Pd—N键的形成[图14(D)~(J)].催化剂被应用于肉桂醛选择性加氢反应中,在温和的压力和温度下达到了较高的催化效果,优于大部分已报道的催化剂体系.
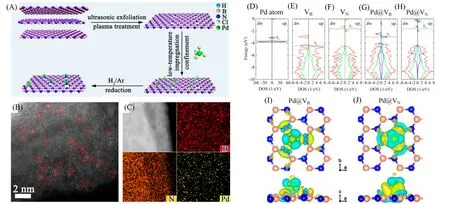
Fig.14 Synthetic route for the construction of Pd1/h⁃BN(A),AC HAADF⁃STEM image(B),energy⁃disper⁃sive X⁃ray elemental mapping(C),spin up and spin down density of states for Pd atom(D),h⁃BN with VB(E),h⁃BN with VN(F),Pd adsorbed h⁃BN with VB(G),and Pd adsorbed h⁃BN with VN(H),top view and side view of difference charge density of Pd adsorbed h⁃BN with VB(I),and Pd adsorbed h⁃BN with VN(J)[86]
总之,在实验和理论层面均验证了超低温液相合成策略的可行性,证实了超低温度反应环境显著影响反应过程中的形核与生长行为的热力学与动力学,普适地合成了可应用于各类催化反应的多种负载型金属单原子催化剂,阐明了低温抑制原子级金属催化剂形核的物理化学机理,并对非均相形核进行了研究,对传统形核理论提出了补充,同时也为大规模、高效合成单原子催化剂开辟了新思路,对于单原子催化的产业化具有重要意义.
4 总结与展望
总结了单原子材料的冷冻合成策略,该策略从反应动力学与热力学的原理出发,抑制反应中粒子的形核与生长行为,普适地在各类载体表面合成了多种金属单原子催化剂,并探究了原子级分散催化剂的形成与锚定机理.通过该方法的应用和理论研究,单原子材料的湿化学合成取得了很大的进展和突破.除催化剂的设计之外,研究人员均对单原子催化剂进行了系统表征,验证了其可应用于各类催化反应,且均表现出优异的催化选择性、活性和稳定性,并对其对应的催化机理进行了深入的探讨,为高性能催化剂的设计奠定了理论基础.虽然单原子催化剂目前已经在诸多领域已经取得了长足的发展,但从基础研究到工业应用还需要开展很多研究工作,目前以下挑战仍亟待解决:
(1)尽管已经开发了多种合成策略,其中一些甚至可以操纵和排列原子,但大批量合成单原子催化剂仍然是一个很大的挑战,如何精准可控地调节单原子配位结构也是一大难题,单原子的高负载量和高表面能导致的团聚之间的本征矛盾也使得高负载单原子的合成显得尤为困难.本文中主要评述的冷冻合成策略也仍存在缺陷,如合成过程中溶液金属组分前驱体的浓度需要保持在较低的水平,以及低温环境下缓慢的反应动力学,为基于该策略大规模合成单原子材料带来挑战.因此,尝试进一步扩大冷冻合成策略的可适用条件范围,并提高合成效率对单原子材料的广泛应用具有重要意义.
(2)在单原子催化剂的表征方面,得益于科研人员的不断努力和表征技术的不断发展,多种先进表征手段已应用于单原子催化剂的探索.然而,目前单原子催化剂的表征严重依赖于EXAFS 和STEM,但STEM仅能反映局部信息,而EXAFS在单原子结构方面的敏感度偏低,团簇催化剂也会表现出与单原子的谱图信息相近的结果.因此,发展更加先进的(如原位表征等)、直接监测反应过程的技术手段将更加有利于对生长机制与催化行为机制的深入理解.
(3)在实际应用中,对于某些活化过程需要多原子协同的反应,单原子催化剂很可能作用有限.这就需要设计多个原子协同的催化活性中心,然而双原子催化剂和三原子催化剂的相关研究仍处于起步阶段,有待进一步深入了解它们的结构和催化性质.此外,单原子材料在催化过程中的动态稳定性也值得深入研究.相对于常规环境,在电解液中容易出现金属原子溶解或表面氧化等问题,而应用环境中单原子的稳定性对于催化剂的设计具有相当重要的意义.
(4)在结构-性能本征关系的热力学、动力学基础研究中,由于宏观性能严重依赖于催化剂的本征电子结构(如d 带位置、功函数和费米能级等),本征结构-性能之间内在关系的深入研究仍然很有必要.尽管单原子催化剂由于结构简单明确、体系较小,容易建构结构模型并进行精确计算,使理论计算与实验的结合验证变得相对容易、准确.但是,中间物的吸附-脱附、反应势垒、表面电荷分布变化和化学键的伸缩振动仍不清晰,在原子层面上的活性位点和反应动力学机理上,目前仍需要更加深入和系统的研究.
猜你喜欢
上海金属(2022年4期)2022-08-03 09:52:10
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
当代陕西(2019年6期)2019-04-17 05:04:10
精密成形工程(2018年6期)2018-11-23 08:31:08
材料工程(2017年7期)2017-07-25 11:20:11
无机化学学报(2014年4期)2014-02-28 17:31:08
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
