
钴单原子的双重限域制备策略及高效CO2电还原性能
2022-09-19 06:28:50杨恒攀何传新
高等学校化学学报 2022年9期
吴 玉,李 轩,杨恒攀,何传新
(1.中国科学技术大学化学物理系,合肥微尺度物质科学国家实验室,合肥 230026;2.深圳大学化学与环境工程学院,深圳功能高分子重点实验室,深圳 518071)
随着大气中二氧化碳(CO2)浓度的日益升高,发展可以将CO2转化为其它可利用资源的方法迫在眉睫,其中电催化二氧化碳还原反应(CO2RR)是一种极其有效的方法[1,2].其中,CO2RR 产物之一的一氧化碳(CO)作为向其它高价值产物(如碳氢化合物和醇类)转化的关键中间产物被广泛研究[3].Cui等[4]模仿人类肺部,设计了一种具有高气体渗透性和低水扩散性的三项催化体系,这种体系可以在‒0.6 V的电位下使产物CO的法拉第效率达到94%[4].Liu等[5]制备了四齿卟啉配体螯合功能化金纳米粒子(AuNPs),其在催化CO2RR制CO时,在过电位为340 mV时电流密度提高了近110倍,CO的法拉第效率也高达93%,并且能够保持至少72 h 的稳定性.将CO2还原为CO 是较为经济且有效的方式之一,比转化为高级烃类和醇类有更高的法拉第效率,也有更适合的技术和经济可行性[6,7].之前,研究人员致力于开发以贵金属为活性中心的催化剂[8,9],然而,贵金属价格较高且在地球上的储量较低,限制了它们在催化方面的应用[10,11].因此,非贵金属材料在催化领域备受关注.
CO2RR是动力学缓慢的过程,因此需要高效的催化剂来提高其反应效率.之前,多使用纳米颗粒及其复合材料作为CO2还原电催化剂,然而在催化过程中,其金属利用率低和比质量活性差等特点限制了它们的应用.单原子催化剂(SACs)由于其高催化性能和高金属利用率引起了广泛关注[12].现已开发了一系列[如原子层沉积法(ALD)[13~15]、湿化学法[16~18]、共沉淀法[19]、浸渍化学法[20]、有机金属配合物法[21,22]以及离子交换法[23]等]合成方法.其中,金属有机框架(MOF)衍生策略[24,25]是制备有机金属配合物的一种方法,MOF在热解的时候可以将金属原子固定在框架中,因此可以有效地阻止催化剂金属的团聚.不仅如此,MOF材料种类的多样性也表明其是具有潜在价值的催化剂合成前驱体.Jiang等[26]利用MOF 网格中配体及金属混合合成的单原子催化剂,有效地阻止了Fe 在碳化过程中的团聚.Li等[27]通过热解和酸刻蚀将UiO-NH2孔道中的Ru金属离子成功限域在N-C材料上,形成Ru单原子催化剂.Zhu等[28]通过对Zn-Fe沸石咪唑骨架高温热化学反应,构建了具有Fe-Nx活性中心的多介孔碳纳米框架.因此,利用MOF衍生的方法来合成单原子催化剂是非常有前景的方法.
沸石咪唑骨架(ZIF)作为一种MOF,因为其具有特定的四面体型三维网状结构被广泛用于合成催化剂材料.本文先将钴(Co)加入到ZIF-8的孔道中形成了ZnCo-ZIF,再通过静电纺丝将ZnCo-ZIF与聚丙烯腈(PAN)共纺丝,将ZIF固定在多孔碳纤维的表面,经过高温热化学反应后,材料中的Zn与PAN挥发形成了一种由壁孔结构和碳纳米纤维双重限域的Co 单原子催化剂(A-Co@PCFs),研究了A-Co@PCFs电催化CO2还原生成CO的性能.
1 实验部分
1.1 试剂与仪器
六水合硝酸锌[Zn(NO3)2·6H2O]、六水合硝酸钴[Co(NO3)2·6H2O]、2-甲基咪唑(2-Methylimidazole)和N,N-二甲基甲酰胺(N,N-Dimethylformamide,DMF)均为分析纯,购自上海阿拉丁生化科技股份有限公司;聚丙烯腈(Polyacrylonitrile,PAN,分析纯)购自西格玛奥德里奇(上海)贸易有限公司;超纯水(电阻率>18 MΩ·cm)由美国Millipore超纯水机制备.
CHI 660c 型电化学工作站,上海辰华仪器有限公司;GenPure UV-TOC 型超纯水机,美国Thermo Fisher Scientific 公司;TL-Pro 型静电纺丝机,深圳市通力微纳科技有限公司;JSM-7800F &TEAM Octane Plus型场发射电子扫描显微镜(FESEM),日本电子公司;JEM-ARF200F TEM/STEM型高角度环形暗场扫描透射电子显微镜(HAADF-STEM)和F200型透射电子显微镜(TEM),搭载了200 kV 加速电压,日本JEOL 公司;Empyrean 型粉末X 射线衍射(XRD),CuKα射线源(λ=0.15406 nm),荷兰PANalytical 公司;Micromeritics BELSORP-max 型氮气吸附-脱附测试仪,日本麦奇克拜尔公司;SRI 8610C 型气相色谱仪(GC,美国SRI 公司);AVANCE Ⅲ600 MHz 超导核磁共振波谱仪(NMR,瑞士Bruker公司);VG Scientific ESCALAB 250型X射线光电子能谱(XPS),美国Thermo公司;Nexion 300电感耦合等离子体质谱仪(ICP-MS),美国Perkin Elmer 公司;X 射线吸收精细结构(XAFS,Co K-edge)光谱仪,Argonne国家实验室先进光子源(APS)12 BM站,储存环在1.85 GeV的正电子下工作,最大束流强度为300 mA,并采用双晶体硅(311)单色仪进行单色同步辐射,在环境条件下,利用电离室荧光模式记录XAFS数据,并使用Athena和Artemis软件包进行分析.
1.2 实验过程
1.2.1 ZnCo-ZIF的合成 将0.056 g Co(NO3)2·6H2O和1.110 g Zn(NO3)2·6H2O溶解在8 mL 去离子水中并充分溶解,记为溶液A;将22.7 g 2-甲基咪唑粉末溶解在80 mL 去离子水中,记为溶液B,然后在磁力搅拌下将溶液A 倒入溶液B 中,室温下搅拌混合5 min 后,将其在10000 r/min 条件下离心40 min(每10 min一次,重复4次),离心完成后倒去上层清液,然后用去离子水充分清洗,重复离心、清洗步骤3次后,收集固体物质,并在60 ℃的真空干燥箱中稳定超过8 h,可得到约80 nm粒径的ZnCo-ZIF纳米颗粒.
1.2.2 ZIF-8 的合成 将1.170 g Zn(NO3)2·6H2O 充分溶解在8 mL 去离子水中,记为溶液C;将22.7 g 2-甲基咪唑粉末加入到80 mL去离子水中,充分溶解后得到溶液D;然后在磁力搅拌条件下迅速将溶液C倒入溶液D中并搅拌5 min.采取与1.2.1节相同的离心-清洗步骤,最后将所得固体物放于真空干燥箱中超过8 h,即可得到粒径约80 nm的ZIF-8.
1.2.3 A-Co@PCF 的合成 将1.2 g ZnCo-ZIF 分散于15 mL 去离子中,在常温下超声1 h 以分散完全.然后将其转移到50 ℃下油浴中,并逐步加入1.2 g PAN,充分搅拌1 h使其均匀分散.然后将分散液在常温下搅拌20 h以上,使其保持均匀稳定的前驱体分散液状态.
将分散均匀的前驱体溶液转移到20 mL纺丝注射器中,然后通过聚四氟乙烯软管连接含有前驱体溶液的注射器与纺丝针头进行静电纺丝.其中工作电压为18 kV,纺丝纤维接收装置与喷射针头的间隔为15 cm,注射速度控制为1.0 mL/h.纺丝完成后将得到的纺丝纤维在真空干燥箱中充分干燥.待充分干燥后,将纺丝纤维放在大小合适的瓷舟中并置于管式炉中,通过温控程序的调整将纺丝纤维分别在800,900和1000 ℃的温度下焙烧1 h,温度控制程序为5 ℃/min,煅烧气氛为氩气(Ar),在1000 ℃保温1 h后自然降温,即可得到高石墨化程度的A-Co@PCFs.
1.2.4 Co NPs@PCFs 和纯PCFs 的合成 将1.2 g ZIF-8 与0.056 g Co(NO3)2·6H2O 在超声下分散到15 mL DMF 中,再以与1.2.3 节相同的条件下加入1.2 g PAN,并在50 ℃油浴条件下搅拌1 h,然后在室温下搅拌20 h 以得到均匀的前驱体分散液.为了保证实验过程中避免不必要的误差,所有的纺丝程序和高温碳化程序均与合成A-Co@PCFs 一致,由此得到的即为Co NPs@PCFs.同样,在合成Co NPs@PCFs过程中,在不加入Co(NO3)2·6H2O情况下,其它步骤不变,即可得到纯PCFs(Pure PCFs).
1.3 电极的制备
将5 mg 样品分散到80 μL Nafion(5%,质量分数)和920 μL 乙醇的混合溶液中制成催化剂墨水.然后将80 μL 准备好的墨水逐步滴加到1 cm2的碳纸上,自然干燥后即可得到测试电极.
1.4 法拉第效率的计算
法拉第效率(FE)计算公式为FE=2F×nCO/(It)[式中:F(96485 C/mol)为法拉第常数;nCO(mol)为产物的摩尔数;I(A)为电流;t(s)为时间].
2 结果与讨论
2.1 材料的合成与表征
A-Co@PCFs的合成步骤如Scheme 1(A)所示,将锌盐、钴盐和2-甲基咪唑在水溶液中混合,并经过离心、洗涤及烘干等步骤得到ZnCo-ZIF,然后将ZnCo-ZIF与PAN进行共纺丝,将得到的初生纤维经过干燥和高温热化学反应即可得到A-Co@PCFs,在此过程中,钴(Co)离子先被合成在ZnCo-ZIF中,在静电纺丝时,包含了Co的ZnCo-ZIF被固定在纺丝得到的纤维上,在高温煅烧时,随着温度的升高锌模块(Zn)逐渐被热解挥发,并最终留下了跟ZIF粒径大约相同的孔道,同时Co也被还原成单原子并被固定在孔道中.详细的合成步骤见1.2.3节.此外,通过将钴盐、ZIF-8和PAN 共纺丝再经干燥和高温煅烧得到Co NPs@PCFs,制备过程如Scheme 1(B)所示.

Scheme 1 Syntheses of A⁃Co@PCFs(A)and Co NPs@PCFs(B)
为了得到样品的形貌信息,采用FESEM 和TEM 进行表征.可见,ZIF-8[图1(A)]和ZnCo-ZIF[图1(B)]有着相似的形貌特征,均为粒径约100 nm的颗粒,而ZIF-8的形状较ZnCo-ZIF明显更加规则和均匀.尽管ZIF 作为前驱体其形貌略有差别,但是利用其纺丝后得到的初生纤维的形貌却几乎相同,都呈现出以表面粗糙的纤维组成的网状结构.这是因为即使ZIF 略有差别,但是在纺丝过程中,ZIF会被包埋在纤维的表面和内部,因此,在纤维表面并不能发现明显的差别[图1(C)和(D)].

Fig.1 FESEM images of ZIF⁃8(A),ZnCo⁃ZIF(B) and pre⁃spinning carbon fibers A⁃Co@PCFs(C)and Co NPs@PCFs(D)
初生纤维在干燥后均进行了高温热化学反应,在高温煅烧过程中Co金属被还原,而ZIF的框架和其中的Zn模块会在高温下挥发,从而形成大量的孔[图2(A)和(B)],而这种多级孔的结构不仅有利于活性中心与电解液的充分接触,也有利于生成气体的脱附.为了探究A-Co@PCFs表面的元素分布,进行了X射线能谱(EDX)元素扫描测试,从图2(C)可以看出,C,N和Co元素都均匀分布在纤维的表面,这说明在合成过程中,Co 元素很均匀地分散于纤维中.通过HAADF-STEM 进一步证实,Co 单原子也很好地分散在纤维上[图2(D)].

Fig.2 FESEM(A) and TEM(B) images of A⁃Co@PCFs,EDX elemental mapping(C) of C,N and Co,HAADF⁃STEM image of A⁃Co@PCFs(D)
A-Co@PCFs[图3(A)],Co NPs@PCFs[图3(B)]和Pure PCFs[图3(C)和(D)]均具有多孔的碳纤维结构,并且纤维直径在500 nm左右;由图3(B)可见,Co NPs@PCFs中Co纳米粒子明显地存在于纤维表面,而其它两种样品表面并未发现明显的纳米颗粒.

Fig.3 FESEM images of A⁃Co@PCFs(A)and pure PCFs(C),TEM images of Co NPs@PCFs(B)and pure PCFs(D)
由FESEM和TEM照片可以看出,所有的纤维均具有多孔结构,因此进一步利用氮气吸附-脱附等温线来探究了纤维材料的比表面积和孔径分布等特性.由图4(A)~(C)可以看出,所有材料均有着相似的Ⅳ类吸附-脱附等温线,表明材料中含有一定量的微孔.同时从孔径分布图可以看出,3种材料的孔结构均以介孔和大孔为主[图4(D)~(F)],其中,A-Co@PCFs相较于其它两种材料有着较多的介孔.由于A⁃Co@PCFs,Co NPs@PCFs和Pure PCFs 3种材料的造孔材料大致相同,因此它们有着相似的比表面积,分别为567,587和603 m2/g.
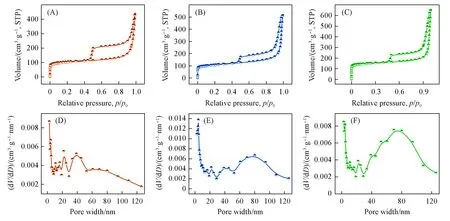
Fig.4 N2 adsorption⁃desorption isotherms(A—C) and pore diameter distribution(D—F) of A⁃Co@PCFs(A,D),Co NPs@PCFs(B,E)and pure PCFs(C,F)
为了进一步探究3种催化材料的结构和性质,进行了XRD和XPS分析.由图5(A)可见,3种样品均没有观察到Co 化合物的峰,而两个分别位于24.1°和44.1°处的峰分别对应的是碳的(002)和(100)晶面,表明3种催化剂材料都具有一定的石墨化程度[29,30].A-Co@PCFs的XRD谱图没有Co的峰,一方面是因为不存在团聚的纳米颗粒,另一方面是因为所有的单原子均被固定在孔道内部;而Co NPs@PCFs 没有Co的峰,归因于Co纳米粒子含量低且基本被包埋在孔道内部.从样品A-Co@PCFs和Co NPs@PCFs 的Co2pXPS 谱可以看出,A-Co@PCFs 的峰位于778.5 和780.5 eV 之间,而Co NPs@PCFs 的峰大多位于778.5 eV[图5(B)],这表明在A-Co@PCFs 中Co 的价态介于0~+2 价(Co-Nx)之间.通过A-Co@PCFs 的N1sXPS 谱可以看出,N1s被分为分别位于401.1,399.8,393.3 和398.4 eV 处的4 个峰[图5(C)],分别对应石墨氮、吡咯氮、Co-N 峰和吡啶氮[31,32],表明N 与Co 和C 耦合,并且成功掺杂到碳基体中.而Co NPs PCFs 的N1sXPS 谱只包含吡啶氮、吡咯氮和石墨氮[图5(D)],表明Co并未与N结合.
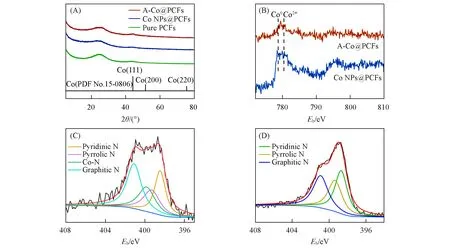
Fig.5 XRD patterns of samples(A),Co2p XPS spectra of of A⁃Co@PCFs and Co NPs@PCFs(B)and N1s XPS spectra of A⁃Co@PCFs(C)and Co NPs@PCFs(D)
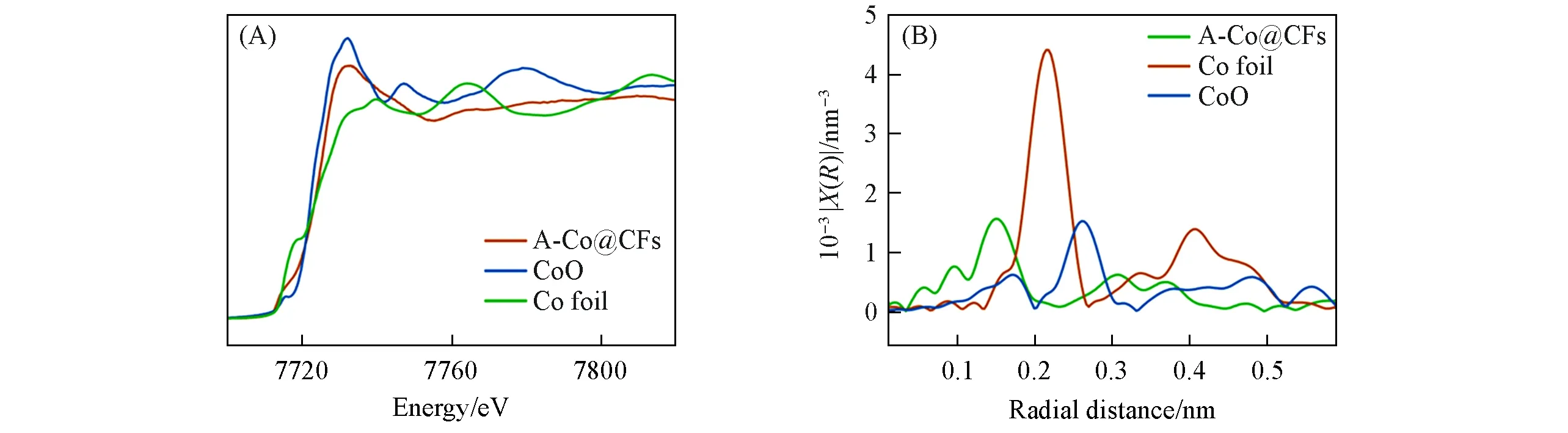
Fig.6 XANES(A)and EXAFS(B)of A⁃Co@PCFs,CoO and Co foil
为了进一步探究Co元素的化学环境,采用XAFs来研究其化学结构.图6(A)是样品A-Co@PCFs、CoO 和Co 箔的X 射线吸收近边结构(XANES)图,由图可知,A-Co@PCFs 的吸收曲线在CoO 和Co 箔之间,表明A-Co@PCFs中Co的价态介于0~+2之间,这与图5(B)的描述一致.扩展X射线吸收精细结构(EXAFS)显示,在0.14 nm 处有明显的Co-N 配位[图6(B)],与A-Co@PCFs 的N1sXPS 谱[图5(C)]相符.
2.2 电催化CO2还原性能
电化学测试在H型电池中利用三电极体系进行,Pt和Ag/AgCl 分别用作对电极和参比电极,制备的电极作为工作电极.采用三电极体系,分别在N2气饱和与CO2饱和的0.1 mol/L KHCO3溶液中测试A-Co@PCFs 电催化CO2RR性能.在进行所有测试之前均用循环伏安(CV)法对电极进行活化.线性扫描伏安(LSV)曲线表明,3种催化剂材料均有一定的催化性能(图7),并且可以看出A-Co@PCFs的电流密度在所有电位下均远高于Co NPs@PCFs 和Pure PCFs[图7(A)].而且对比在N2气和CO2饱和的电解液中的表现可知,材料Co NPs@PCFs 和Pure PCFs 催化CO2RR 过程电流密度较低[图7(C)和(D)],而A-Co@PCFs 具有更好的催化CO2RR 性能[图7(B)].A-Co@PCFs 在催化过程中在‒0.66 V(vs.RHE)时电流密度达到26 mA/cm2,而另外两种材料在该电位下的电流密度均在5 mA/cm2以下[图7(A)].
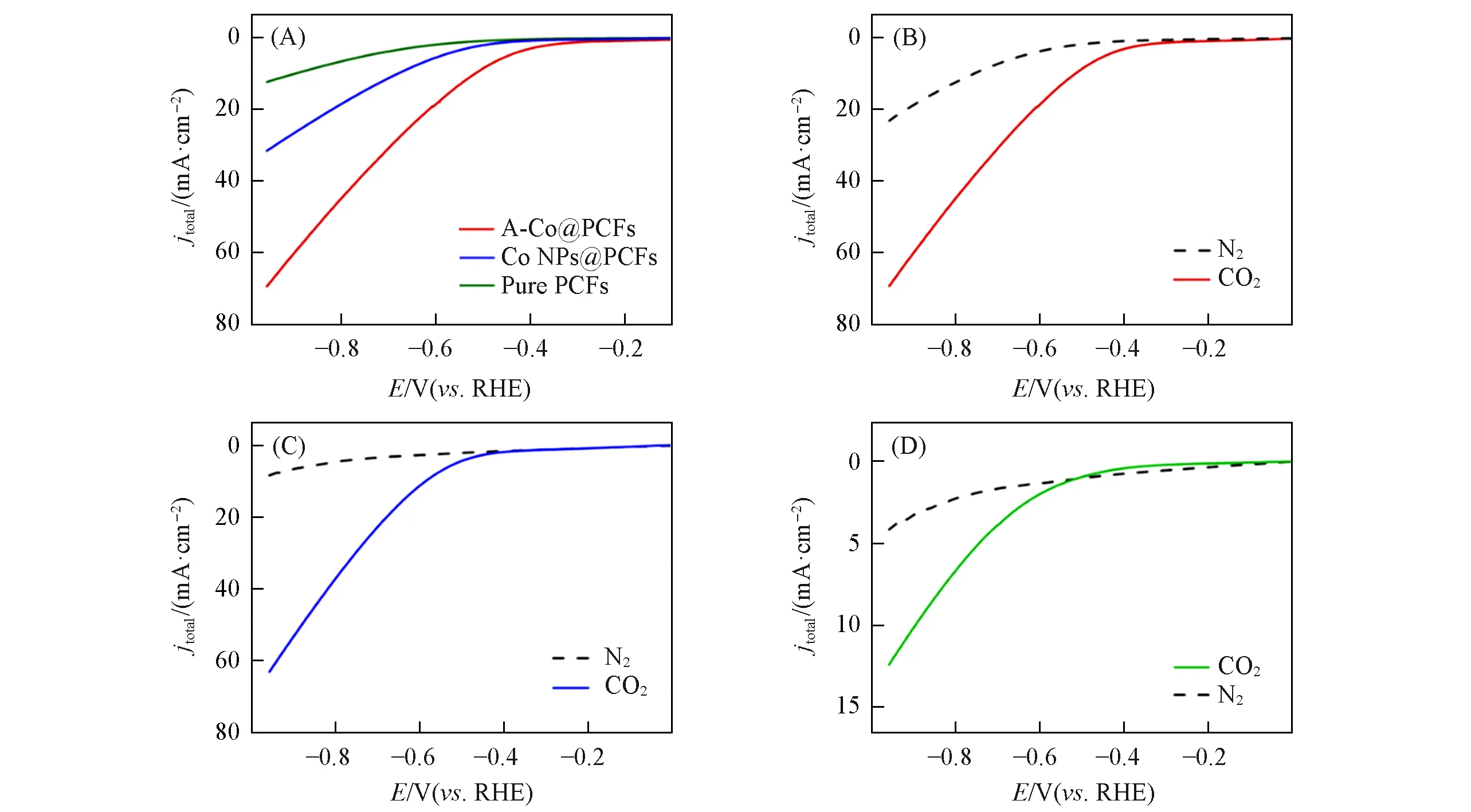
Fig.7 LSV curves of three samples(A),LSV curves of A⁃Co@PCFs(B),Co NPs@PCFs(C)and pure PCFs(D)in N2⁃and CO2⁃saturated 0.1 mol/L KHCO3
催化CO2RR 得到的气体产物和液体产物分别用气相色谱仪(GC)和核磁共振氢谱(1H NMR)进行测定.在各个电位下进行长时间的电解反应,收集产物,1H NMR测试得到的数据如图8(A)所示,3种催化剂在所有化学位移下均没有产生任何液体产物[取自‒0.66 V(vs.RHE)时的电解产物],并且CO是催化CO2RR 的唯一气体产物.经对气相产物的测试和生成气体的法拉第效率计算,得知3 种材料都具有一定的催化性能,其中,A-Co@PCFs 在催化CO2RR 时在所有测试电位下均能达到超过40%的CO法拉第效率,其中,在‒0.66 V(vs.RHE)时能够达到最大约94%的CO法拉第效率[图8(B)];材料Co NPs@PCFs 既能在催化CO2RR 时有一定的性能(所有测试电位下的CO 法拉第效率均不超过55%),也能在催化H2气析出时有一定的性能[图8(C)],因此其并不具有良好的选择性,而Pure PCFs在所有测试电位下都有大量氢气析出(CO的法拉第效率在所有电位下均不超过10%)[图8(D)].
不仅如此,A-Co@PCFs 在‒0.26~‒0.96 V(vs.RHE)电位范围中,生成CO 的法拉第效率先升高再下降,并在‒0.66 V(vs.RHE)时达到94%,而其它两种催化剂的产生CO 法拉第效率分别为33%(Co NPs@PCFs)和5.4%(Pure PCFs),而且在测试的所有电位下,其产生CO法拉第效率均高于另外两种催化剂[图9(A)].由于反应产物只有CO和H2,因此产生H2的法拉第效率与产生CO的法拉第效率在数值上互补[图9(B)].

Fig.8 1H NMR spectra of the electrolyte after 4 h CO2 reduction electrolysis at -0.66 V(vs.RHE) for three samples(A),CO and H2 Faradaic efficiencies of A⁃Co@PCFs(B),Co NPs@PCFs(C)and pure PCFs(D)
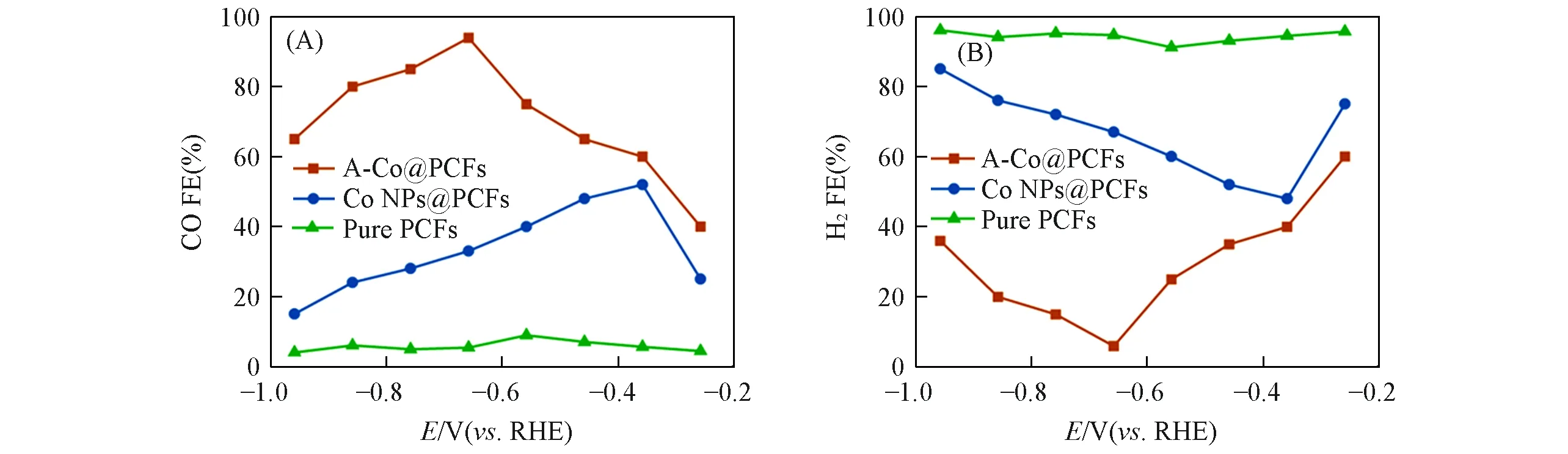
Fig.9 CO(A)and H2(B)Faradaic efficiencies at different potentials for three samples
为了进一步衡量样品的催化效率,计算了Co活性位点催化产生CO的转化频率(TOF)值.如图10所示,A-Co@PCFs 的转化频率能分别在‒0.66,‒0.76,‒0.86 和‒0.96 V(vs.RHE)时达到6950,8240,10700,11260 h‒1,相比于Co NPs@PCFs 的转化频率近乎为0 有着极大的优势,这表明A-Co@PCFs具有更好的本征活性.
材料在催化过程中的稳定性优劣是衡量催化材料能否长久稳定使用的重要标准之一,因此对A-Co@PCFs 材料在−0.66 V(vs.RHE)进行了60 h的稳定性测试.由图11(A)可以看出,其产生CO 的法拉第效率并没有明显降低,同时该电位下的电流密度也没有过多地减小,而且在经过60 h电解后,其结构没有发生变化[图11(B)],形貌也没有发生较大的变化[图11(C)和(D)],这都表明该材料在作为CO2RR催化剂时具有优异的稳定性,这也是其优势所在.
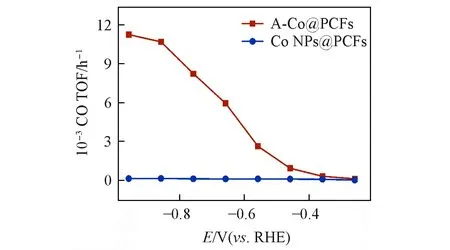
Fig.10 Corresponding CO TOF for A⁃Co@PCFs and Co NPs@PCFs

Fig.11 Long⁃term stability test of A⁃Co@PCFs at-0.66 V(vs.RHE)for 60 h(A),XRD patterns(B),SEM(C)and TEM(D)images of A⁃Co@PCFs after tests
3 结 论
利用MOFs纳米颗粒和碳纳米纤维的双重限域作用,合成了碳纳米纤维负载的钴单原子材料.这种合成方法简单有效,高度分散的钴单原子作为主要活性位点,碳纤维丰富的孔道结构有利于活性位点充分暴露于固/液/气三相界面.A-Co@PCFs 材料的多级孔结构还为反应物的传质、电子的传输及产物的脱附提供了便利.A-Co@PCFs材料在催化CO2RR产生CO时最高能够达到94%的法拉第效率,并且能够保持至少60 h的催化稳定性,表明其是一种优异的催化材料.同时该种合成方法也为其它单原子材料的合成提供了一定的借鉴.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02 02:33:28
云南化工(2021年7期)2021-12-21 07:27:36
食品安全导刊(2021年20期)2021-11-28 00:56:56
小学生必读(高年级版)(2019年3期)2019-06-21 06:50:28
天天爱科学(2017年2期)2017-04-29 00:44:03
电镀与环保(2016年2期)2017-01-20 08:15:26
现代工业经济和信息化(2016年12期)2016-05-17 05:37:52
燕山大学学报(2015年4期)2015-12-25 02:19:46
合成技术及应用(2015年3期)2015-12-11 08:36:27
电源技术(2015年9期)2015-06-05 09:36:06
