
单原子铈对弱芬顿效应活性位点氧还原稳定性的提升
2022-09-19 06:29:02楚宇逸罗二桂刘长鹏葛君杰
高等学校化学学报 2022年9期
楚宇逸,兰 畅,罗二桂,刘长鹏,葛君杰,邢 巍
(1.中国科学院长春应用化学研究所电分析化学国家重点实验室,吉林省低碳化学动力重点实验室,长春 130022;2.中国科学技术大学应用化学与工程学院,合肥 230026;3.山西师范大学化学与材料科学学院,磁性分子与磁信息材料教育部重点实验室,太原 030031)
质子交换膜燃料电池(PEMFC)是一种清洁、高效的能源装置,但阴极所使用的铂族贵金属催化剂存在成本高、资源不足等问题,限制了燃料电池的规模商业化,因此发展非贵金属催化剂是燃料电池未来发展的必经之路[1~5].近年来,基于对d带电子的认知来调控氧还原中间产物的结合能,过渡金属氮掺杂碳(M-N-C)型催化剂的活性越来越接近贵金属铂,成为了最有希望替代铂的材料[6~12].但催化剂的实用化程度也由稳定性所决定,目前,M-N-C型阴极催化剂在实况工作下,工作100 h活性往往衰减了40%~80%,不能满足实际工作的需求,其稳定性亟待提升[2,13~15].
以Fe-N-C 为例的催化剂在酸性电解质中的活性衰减机制包括碳基底的氧化[13,16~20]、催化中心(Fe-Nx位点)金属离子的脱落和溶出[16,20]和配位氮的质子化[21].值得注意的是,含铁的活性位点能通过芬顿反应,催化氧还原过程中的中间产物H2O2,产生活性氧物种(ROS),而这些富有攻击性的ROS(尤其是·OH)将不可逆地破坏碳基底和燃料电池中的质子交换膜.Kumar等[20]证实燃料电池在O2条件下的循环伏安测试后,发生了碳腐蚀,并导致了FeNx氧还原活性位点的损失;Choi等[16]发现Fe-N-C催化剂暴露于H2O2溶液后转化频率(TOF)值变低,认为这是由于碳基底表面的氧化削弱了FeNx位点吸附O2的结合能所致.综上,含铁的活性中心对电池性能的衰减起主要作用,因此,即使Fe-N-C催化剂是目前活性表现最好的催化剂,在燃料电池中也应尽量避免Fe的引入[16,22].近年来,研究者加强了对抗芬顿活性位点的研究.我们课题组[23]开发了一种具有Cr-N4活性位点的氧还原催化剂,其芬顿反应活性比Fe-N-C低了23倍,在20000次测试后,半波电位仅下降15 mV.Xiao等[24]合成了对*OH具备最优吸附能的单原子催化剂Ru-SSC,其表现出优异的活性,并且得益于Ru中心的低芬顿活性,稳定性表现良好(20000次循环后仅衰减17 mV).
在贵金属燃料电池中,为了避免含铁器件的芬顿反应的影响,常引入具有Ce3+/4+氧化还原电对的CeOx颗粒来清除ROS,以提高稳定性[25,26].Xie等[27]使用Ta-TiOx纳米颗粒添加剂,以达到清除自由基保护Fe-N-C催化剂免受损害的目的,但在M-N-C催化剂中,活性位点是原子级分散的,而且活性氧自由基存在的时间短[28],难以在形成并全部扩散到纳米颗粒上后被消除,并且纳米氧化物易与酸反应,一旦发生离子扩散,还会破坏膜的稳定性[29~32].因此,本文提出向Ru-N-C 催化剂中引入单分散稳定的Ce位点的策略,在减少活性中心受芬顿反应影响的同时,清除ROS,保护碳基底;通过一锅法合成催化剂Ru,Ce-N-C,表征了其结构和形貌,测试了其电催化性能,并研究了其催化反应机理.
1 实验部分
1.1 试剂与仪器
Zn(NO3)2·6H2O(纯度99.99%)、2-甲基咪唑(纯度98%)、RuCl3·6H2O(纯度99%)、CeCl3·7H2O(纯度99.99%)和FeCl3·6H2O(纯度99.99%)购于阿拉丁试剂公司;高纯HClO4(质量分数70.0%~72.0%)和Nafion 溶液(5%,质量分数)购于Sigma-Aldrich 公司;商业20%Pt/C 催化剂(20%,质量分数)购于Johnson Matthey 公司;甲醇、无水乙醇和浓盐酸均为分析纯,购于北京化工厂.所用水均为超纯水(Milli⁃Q Advantage仪器,美国Millipore公司,电阻率18.25 MΩ·cm).
XL 30ESEMFEG 场发射扫描电子显微镜(FESEM)和TECNAI G2 透射电子显微镜(TEM,荷兰FEI公司);PW1700 型全自动X 射线衍射仪(XRD,CuKα,荷兰Philips 公司);ASAP 2020 吸附分析仪(BET,美国Micromeritics公司);电感耦合等离子体发射光谱仪(ICP-OES,美国Thermo Fisher Scientific公司);XSAM-800 X 射线光电子能谱仪(XPS,美国Kratos 公司);同步辐射装置(SSRF)BL14W1 站(中国科学院上海应用物理研究所),单色仪(法国HORIBA公司),进行Ru K边和Ce L边X射线吸收精细结构(XAFS)测试;750E型双恒电位仪(CH Instruments公司),电化学测量均在三电极体系中进行.
1.2 样品的制备
采用一锅法制备Ru,Ce-N-C以及对比样Ru-N-C,Fe-N-C和Ce-N-C系列催化剂[33].
首先,将3.25 g 2-甲基咪唑溶解在40 mL 甲醇中,随后加入20 mL 含有1.5 g Zn(NO3)2,6.8 mg RuCl3·3H2O和6 mg CeCl3·7H2O的水溶液,室温搅拌24 h,离心分离,用乙醇洗涤沉淀3次后,于60 ℃下恒温干燥12 h,得到的前驱体命名为Ru,Ce@ZIF-8.依照类似的方法,合成了其它先驱体Ru@ZIF-8,Fe@ZIF-8,Ce@ZIF-8 和纯ZIF-8,区别在于:在上述步骤中除加入1.5 g Zn(NO3)2外,Ru@ZIF-8 需加入6.8 mg RuCl3·3H2O,Fe@ZIF-8 需加入31 mg FeCl3·6H2O,Ce@ZIF-8 需加入14.0 mg CeCl3·7H2O,纯ZIF-8则不加入其它金属盐.
将干燥后的前驱体粉末分别放入石英舟中,在管式炉内Ar气氛中,以5 ℃/min的升温速率加热至950 ℃,恒温1 h后,自然冷却至室温.用稀盐酸酸洗黑色碳化产物12 h,过滤,用足量的去离子水洗涤滤饼后,放入真空干燥箱于60 ℃干燥12 h 后,取出研磨,待用,所得样品分别命名为Ru,Ce-N-C,Ru-N-C,Fe-N-C 和Ce-N-C.对比样xRu,yCe-N-C 和Ru,Ce-N-C-z与Ru,Ce-N-C 的合成方法一致,区别在于:前者前驱体中RuCl3·3H2O和CeCl3·7H2O的加入量分别为Ru,Ce-N-C的x倍和y倍,后者前驱体中RuCl3·3H2O和CeCl3·7H2O的加入量均为Ru,Ce-N-C的z倍.
1.3 XAFS测试与数据分析
SSRF 的电子储能环工作电压为3.5 GeV,最大电流为250 mA.在荧光模式下收集XAFS 数据,分别使用Ru 箔和Fe 箔校准能量.另外,分别使用固定出口双晶Si(311)和Si(111)单色仪收集XAFS 数据.利用IFEFFIT软件包的ATHENA模块,根据标准程序执行获得扩展X射线吸收精细结构(EXAFS)数据[34,35].
1.4 电化学性能测试
将5 mg 催化剂超声分散在含有50 μL Nafion(5%)溶液和950 μL 无水乙醇的悬浮液中制备墨水.取一定量墨水滴涂(采用旋涂的方式)在玻碳旋转圆盘圆环电极(RRDE)上.对于非贵金属催化剂,催化剂负载量均为0.6 mg/cm2,以20%Pt/C催化剂(Pt负载量为30 μg/cm2)作为对比.电极体系以饱和甘汞为参比电极,碳棒为对比电极,旋转圆盘电极(RDE)或RRDE为工作电极.
电解质溶液为0.1 mol/L HClO4溶液,通过线性扫描伏安法(LSV),以10 mV/s 的扫描速率,1600 r/min的转速,在1.1~0.2 V的电位区间进行RDE-ORR(氧还原反应)测试,对于RRDE,环电极电位设为1.3 V.通过下面公式用于计算ORR过程的表观电子转移数(n)和H2O2的产率(y,%).

式中:ID(mA)为盘电流;IR(mA)为环电流;N为环电极对H2O2的收集系数(37%).
所有电位转化为相对于可逆氢电极电位,在酸性介质中ERHE=ESCE+0.304 V;加速老化测试:在O2气饱和的0.1 mol/L HClO4溶液中,在0.6~1.0 V 电位区间内,以200 mV/s 的扫描速率循环伏安扫描30000次.
2 结果与讨论
2.1 催化剂的形貌与结构表征
对合成Ru,Ce-N-C催化剂的前驱体进行了表征.图1(A)为ZIF-8的SEM照片,可见,未引入Ce金属盐前驱体的纯ZIF-8粒径(200 nm)较大,形状为正十二面体;引入Ce盐的Ce@ZIF-8的粒径明显缩小至50 nm[图1(B)];Ru,Ce@ZIF-8的粒径与Ce@ZIF-8相似[图1(C)].图1(D)为催化剂前驱体的XRD谱图,可见,纯ZIF-8结构为具有沸石晶型和有序孔道的高水热稳定性金属有机框架(MOF)材料,各个衍射峰的位置、强度与文献[33]报道一致,且未观察到杂质峰.由于引入的金属盐为痕量,在其它前驱体的XRD谱图中,所有的前驱体结构都与ZIF-8相似,未出现金属盐的晶型.这是部分取代了MOF中的Zn节点所导致,铈金属盐没有在这个过程中形成CeO2,Ce(NO3)3可以在甲醇溶液中与2-甲基咪唑配位,在水中则生成CeO2[36].

Fig.1 SEM images of ZIF⁃8(A),Ce@ZIF⁃8(B)and Ru,Ce@ZIF⁃8(C),XRD patterns of ZIF⁃8,Ce@ZIF⁃8,Ru,Ce@ZIF⁃8 and CeCl3·7H2O(D)
经过热解和酸洗步骤之后,使用ICP-OES表征对样品进行元素分析,Ru,Ce-N-C催化剂中钌含量为0.727%(质量分数),铈含量为0.483%.对比Ru-N-C 催化剂(钌含量为0.241%),发现引入铈金属盐后,催化剂中Ru含量提高了近3倍,这可能是由于Ce节点部分替代了Zn节点,而Ce的离子半径显著高于Zn,导致MOF孔道缩小,能更多地捕获钌盐,使其均匀分散在笼中,避免聚集形成的颗粒在酸洗步骤中被洗去.Ce-N-C中铈的含量为0.117%,低于Ru,Ce-N-C催化剂中的铈含量.Fe-N-C中铁含量为1.649%,与文献[33]报道的含量相似.
由SEM照片发现,热解后的Ru,Ce-N-C催化剂表面收缩明显,这是由于热解过程中,Zn节点以蒸气的方式挥发所致[图2(A)].如TEM 照片所示,Ru,Ce-N-C 催化剂表面没有发现明显的金属颗粒[图2(B)].在热解后的各催化剂的XRD谱图中,只出现了无定型碳的宽峰[图2(C)],并未出现晶型的Ru或Ce物种,说明金属源以原子级分散在催化剂表面的,与TEM结果相符.

Fig.2 SEM image(A)and TEM image(B)of Ru,Ce⁃N⁃C,XRD patterns of the pyrolyzed N⁃C,Ru,Ce⁃N⁃C,Fe⁃N⁃C and Ce⁃N⁃C samples(C)
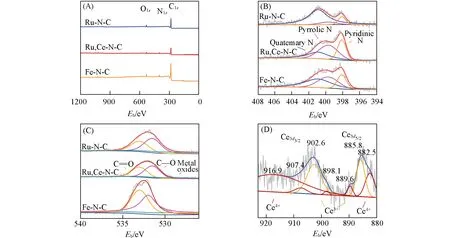
Fig.3 Survey(A)and high⁃resolution XPS spectra of N1s(B)and O1s(C)for Ru,Ce⁃N⁃C,Ru⁃N⁃C and Fe⁃N⁃C,high⁃resolution XPS spectra of Ce3d of Ru,Ce⁃N⁃C(D)
为了探究Ru,Ce-N-C 中Ru 位点和Ce 位点的存在形式,使用XPS 对样品进行表征.图3(A)为Ru,Ce-N-C 的XPS 全谱图,可以看到明显的O1s,N1s和C1s峰.N1s的XPS 谱证实了掺杂氮存在吡啶氮(398.1 eV)、吡咯氮(399.7 eV)和石墨氮(400.9 eV)3 种形式[图3(B)].Ru,Ce-N-C 和Fe-N-C 中以吡咯氮和吡啶氮的存在形式为主,这两种氮已被证明为M-N-C催化剂中活性位点金属配位氮的主要存在形式[22].O1s的XPS 谱表明样品中的氧主要与碳进行配位[图3(C)],金属氧化物的含量很少.Ce3d的XPS谱证实了Ru,Ce-N-C中同时存在Ce3+和Ce4+[图3(D)].此外,由于Ru3d特征峰位置(Eb=280.7 eV)与C1s(Eb=284.5 eV)过于接近,因此,通过Ru,Ce-N-C缺乏Ru3p峰证明催化剂中Ru含量很低(图S1,见本文支持信息),推测Ru在其中以原子级大小存在.
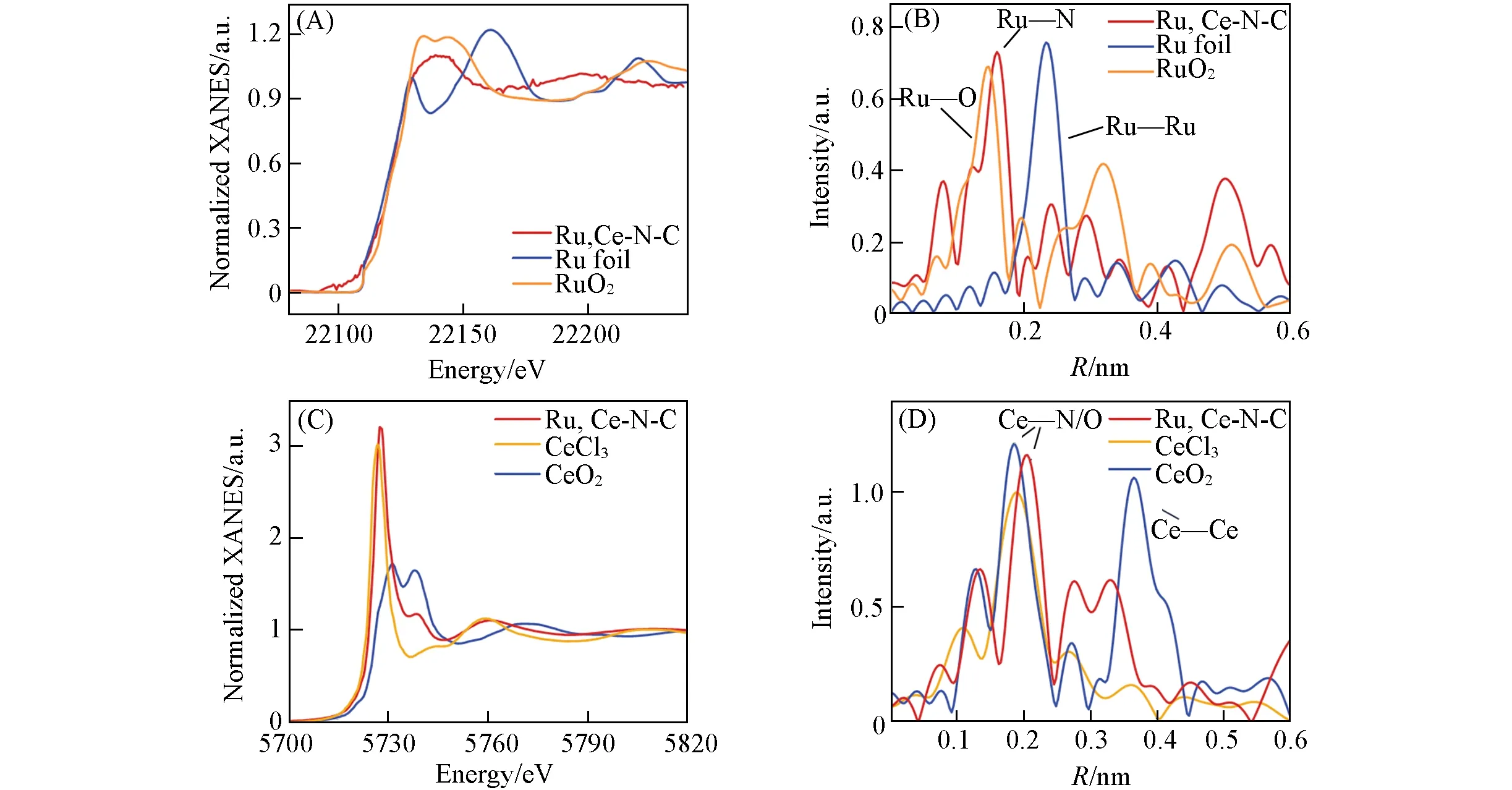
Fig.4 Ru K⁃edge XANES spectra(A),Fourier transforms of k2⁃weighted Ru K⁃edge EXAFS spectra(B),Ce L⁃edge XANES spectra(C),Fourier transforms of k3⁃weighted Ce L⁃edge EXAFS spectra(D)for Ru,Ce⁃N⁃C and standard samples
在X 射线吸收近边结构谱(XANES)中,Ru,Ce-N-C 的峰强度强于Ru 箔[图4(A)],这说明Ru在Ru,Ce-N-C 和Ru-N-C 中的价态(0~+4)更高.在经过傅里叶变换的扩展X 射线吸收精细结构谱(FT-EXAFS)中,Ru,Ce-N-C 在0.16 nm 处显著的峰归为Ru—N 键[图4(B)].同时,并没有发现金属Ru—Ru(0.25 nm)键和Ru—O键(0.14 nm),说明Ru,Ce-N-C中没有Ru金属颗粒和氧化物,Ru的存在形式以单位点RuNx为主,与XPS结果相符(图S1).同样,对Ru,Ce-N-C中的Ce位点进行了结构分析.在XANES 谱中,Ru,Ce-N-C 的峰强度位于+3 价的CeCl3和+4 价的CeO2之间[图4(C)],这说明该CeNx位点价态在+3~+4 之间.由图4(D)可见,Ce L 边的EXAFS 谱中表现出与CeO2完全不同的键长,与文献[37]中的CeNx结构谱类似.以上表征结果说明,在Ru,Ce-N-C 催化剂中存在两种单分散的RuNx和CeNx位点.
2.2 催化剂的电催化性能
对制备的催化剂进行了电化学ORR 性能测试.利用RRDE,在1600 r/min 电极转速下,在O2气饱和的0.1 mol/L HClO4电解液中,进行LSV测试,并与商业20%Pt/C催化剂进行性能对比.首先,通过测试850,950和1050 ℃热解温度下制备的催化剂[图5(A)],发现在950 ℃下,得到的Ru,Ce-N-C性能最优,而850 ℃热解后几乎没有性能.这是由于不同温度提供的能量不同,金属前驱体的分散程度不同,对最终的活性位点分散程度有着决定性的影响[38].因此,为了最大化获得单分散位点的催化剂,均以950 ℃热解温度制得的催化剂进行研究.

Fig.5 Comparison of ORR polarization curves of Ru,Ce⁃N⁃C catalysts during different pyrolyzed tempera⁃tures(A),comparison of ORR polarization curves of Ru,Ce⁃N⁃C catalysts with different additions of RuCl3·3H2O(B),CeCl3·7H2O(C),RuCl3·3H2O and CeCl3·7H2O(D) in precursors,comparison of ORR polarization curves of Ru,Ce⁃N⁃C,N⁃C,Ru⁃N⁃C,Pt/C and Ce⁃N⁃C catalysts(E),the H2O2 yield and electron⁃transfer number(n)on different catalysts determined by RRDE technique(F)
如图5(B)所示,保持铈盐加入量不变,对前驱体中的钌盐加入量进行优化.当减少Ru 的加入量时,由于活性位点密度减少,催化剂活性降低.而增加Ru的加入量,易发生水解团聚形成颗粒,催化剂单分散的有效位点数量减少,活性也会降低.保持钌盐加入量不变,对前驱体中的铈盐加入量进行优化,发现随着铈盐加入量的增加性能反而下降[图5(C)],这可能是由于铈盐量增加后,反而聚集形成颗粒,使得对Ru位点的锚定效果降低.对金属盐的整体加入量扩大或缩减[图5(D)],催化剂的活性与前驱体引入量呈火山型关系,太少位点密度不够,太多则容易团聚形成颗粒,导致活性下降.其中,Ru,Ce-N-C-1.0催化剂(即Ru,Ce-N-C)性能最好,半波电位为0.78 V;活性越高,H2O2产率越低,最佳催化剂的H2O2产率约为3.8%.Ru,Ce-N-C 催化剂的性能不仅优于用同样方法制备的Ru-N-C 和Fe-N-C 催化剂[图5(E)],并且其H2O2的产率大幅降低.虽然Ce-N-C 催化剂无ORR 活性[图5(C),仅略好于N-C],但其H2O2产率远小于N-C催化剂[图5(F)],说明CeNx位点对H2O2具有降解作用.
2.3 催化剂的稳定性
稳定性是衡量催化剂的重要指标,在O2气饱和条件下,对Ru,Ce-N-C 催化剂进行了加速老化测试,并与Fe-N-C 催化剂进行比较,即在0.6~1.0 V 电位区间以200 mV/s 进行循环伏安(CV)扫描,比较老化前后的LSV 曲线.如图6(A)所示,Ru,Ce-N-C 的半波电位在30000 次CV 测试后仅负移了9 mV,远低于Fe-N-C催化剂(半波电位负移60 mV)和商业Pt/C催化剂(半波电位负移48 mV),证实了其优异的稳定性.与近3 年来文献报道的结果相比,其稳定性表现都非常突出(表S1,见本文支持信息)[23,39~42].值得注意的是,与Fe-N-C催化剂在第10000次循环后就明显衰减了20 mV不同[图6(B)],Ru,Ce-N-C 催化剂反而在第10000 次加速老化测试后,活性有明显的增加(正移19 mV),再衰减至与初始相同的性能,最后活性趋于稳定不变.结合ORR电子转移系数的变化[图6(D)],这可能是由于在初始测试时,有部分RuNx位点并没有暴露完全,部分H2O2形成的自由基对碳基底的表面进行了刻蚀,使得活性位点暴露增加,活性升高.
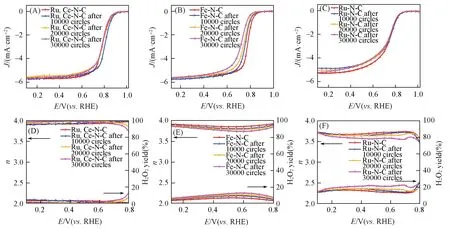
Fig.6 Accelerated degradation test(ADT) of Ru,Ce⁃N⁃C(A),Fe⁃N⁃C(B) and Ru⁃N⁃C(C) catalyst by cycling the potential(0.6—1.0 V) in O2⁃saturated 0.1 mol/L HClO4 for 30000 cycles,the H2O2 yield and elec⁃tron⁃transfer number(n) during ADT on Ru,Ce⁃N⁃C(D),Fe⁃N⁃C(E) and Ru⁃N⁃C(F) determined by RRDE technique
对比老化前后的环盘数据,发现活性损失得越多ORR电子转移数越低,H2O2越高,而这个趋势一旦发生[图6(E)],就会形成不可逆转的结果.为了验证CeNx位点的引入有助于消除自由基,提高催化剂的稳定性,测试了对比样Ru-N-C催化剂的稳定性[图6(C)],其半波电位负移了24 mV,且在这个过程中H2O2产率远高于Ru,Ce-N-C 催化剂.Ru-N-C由于自身RuNx位点的抗芬顿作用,位点本身稳定性高于Fe-N-C 催化剂,但H2O2产率较高[图6(F)]导致的碳腐蚀,仍然使其在30000 次加速老化测试后具有明显的损失,具有自由基消除剂的CeNx位点的Ru,Ce-N-C催化剂的稳定性则更为优异.
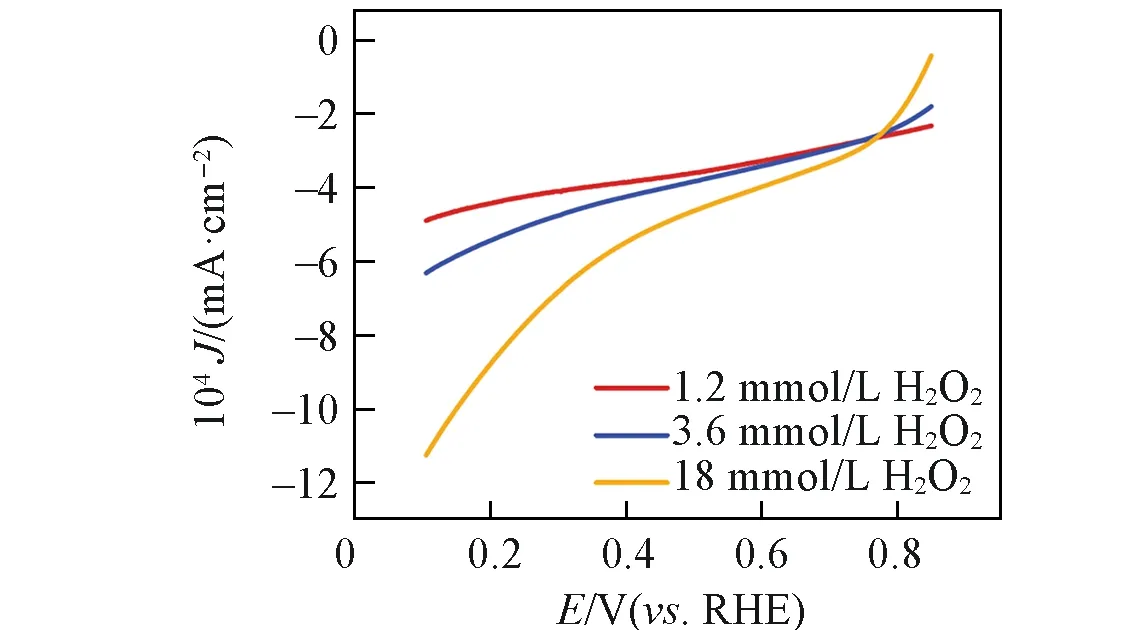
Fig.7 Hydrogen peroxide reduction reaction(HPRR) activity of Ce⁃N⁃C(600 μg/cm2)in N2⁃saturated 0.1 mol/L HClO4
为了探究催化剂高稳定性的来源,对Ce-N-C催化剂进行了H2O2电催化实验(图7).可见,在电化学过程中,CeNx起到的作用并不是还原H2O2,而是在高电位下高效地将H2O2氧化为O2,避免了H2O2在反应过程中的积累,减少了芬顿反应的发生,使得O2在整个过程中能以更高的效率还原为H2O.综上,单位点CeNx对自由基的有效清除和抗芬顿RuNx位点的协同作用,有效地提高了Ru,Ce-N-C催化剂的稳定性.
3 结 论
发展了一种单分散铈位点作为自由基清除剂,并结合抗芬顿钌位点的新型高稳定性非贵金属氧还原催化剂Ru,Ce-N-C.在酸性条件下,Ru,Ce-N-C 催化剂的ORR 性能优异,且催化过程接近理想的四电子过程,能够有效抑制芬顿效应的负面影响.通过结构表征和电化学分析,发现RuNx位点是该催化剂的活性主要来源,而能够有效清除自由基的CeNx位点决定了Ru,Ce-N-C的高稳定性.研究结果有助于加深对M-N-C型催化剂稳定性的理解,并且为通过原子级微观结构设计制备高稳定性的非贵金属氧还原催化剂提供了新的思路.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20220294.
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
生物学通报(2019年3期)2019-02-17 18:03:58
中国资源综合利用(2017年2期)2018-01-22 02:44:58
中国资源综合利用(2017年2期)2018-01-22 02:44:58
材料科学与工程学报(2016年1期)2017-01-15 13:33:52
环境科技(2016年3期)2016-11-08 12:14:14
当代化工研究(2016年7期)2016-03-20 16:21:54
中国资源综合利用(2016年11期)2016-01-22 02:01:28
电源技术(2015年9期)2015-06-05 09:36:06
