
S掺杂Fe-N-C高活性氧还原反应催化剂
2022-09-19 06:28杨博龙吴文依向中华
高等学校化学学报 2022年9期
程 前,杨博龙,吴文依,向中华
(北京化工大学有机无机复合材料国家重点实验室,北京 100029)
质子交换膜燃料电池(PEMFC)具有高效节能、原料来源广泛和环境友好等优点,是目前最有前景的能量转换装置之一[1~3].控制燃料电池能量转化效率的关键在于阴极氧还原反应(ORR),目前,商用贵金属铂基催化剂存在价格高昂和储量贫乏等问题,极大限制了PEMFC 的应用[4,5].因此,开发低成本、高催化活性和优良稳定性的非贵金属氧还原催化剂是PEMFC发展的关键.
杂原子掺杂的过渡金属碳基催化剂(M-N-C)价格低廉、活性高且耐久性好,引起了研究者们的关注[6~17].以Fe-N-C 型催化剂为代表,氮原子掺杂后以吡啶态N、Fe-Nx、吡咯态N、石墨态N 和氧化态N[18]的形式存在,其中吡啶态N可调整局部Fe-N-C的活性电子结构,石墨态N可加速氧还原过程中的电子转移,对ORR 催化活性有重要作用[19].同时,调控Fe 含量可以在催化剂表面形成碳包裹的Fe/Fe3C 核壳结构,同样可作为ORR 催化的活性位点.Sun 等[20]发现原子级分散的FeN4活性点与Fe/Fe3C纳米复合结构共存时,两者之间的偶联效应有利于增强催化剂材料的ORR性能.而S原子的共掺杂可以进一步提升ORR活性.Zheng等[21]证明S原子的掺杂使样品中吡啶N、Fe-Nx和石墨N含量增多,且噻吩硫结构改变了碳骨架的电子结构,降低了过电位,有利于氧还原反应的发生.由金属离子或离子簇与有机配体经由配位作用,自组装形成的金属-有机骨架(Metal organic frameworks,MOFs)具有高比表面积、长程有序结构、孔径可调和形貌可控等优点.作为MOFs 材料的一类,沸石咪唑骨架(ZIFs)通过过渡金属离子与咪唑单元之间的配位作用形成,且结构规整有序、三维稳定,经常在制备过程中作为模板来调控催化剂的形貌结构[22].其中,ZIF-8 材料可以通过Zn2+与2-甲基咪唑的配位合成,且高温热解过程中沸点较低的Zn易挥发,并得到多孔碳骨架,有利于催化剂中活性位点暴露和质量传递.因此,对ZIFs 材料进行杂原子掺杂与有效的后处理等方法,可以设计制备出高催化活性的ORR电催化剂[23,24].
本文采用微波加热和高温碳化技术,以ZIF-8 为前驱体,在甲醇-水双溶剂体系中先后引入Fe(NO3)3·9H2O 和KSCN,通过调控Fe,S 的掺杂量,制备得到具有高比表面积和高孔隙率的Fe3C/Fe-SAS@SNC氧还原催化剂;在0.1 mol/L KOH电解液中,Fe3C/Fe-SAS@SNC表现出了优异的氧还原性能,其半波电位达到了0.880 V(vs.RHE),稳定性也优于商业Pt/C催化剂;该材料在酸性环境下也表现出良好的氧还原性能(半波电位0.785 V,vs.RHE),是一种极具发展潜力的非贵金属氧还原催化剂.
1 实验部分
1.1 试剂与仪器
六水合硝酸锌[Zn(NO3)2·6H2O]和硫氰化钾(KSCN)均为分析纯,上海阿拉丁生化科技有限公司;九水合硝酸铁[Fe(NO3)3·9H2O]、氢氧化钾(KOH)和2-甲基咪唑(2-MeIm)均为分析纯,上海麦克林生化科技有限公司;无水甲醇、无水乙醇和硫酸均为分析纯,北京化工厂;杜邦膜溶液(Nafion,质量分数5%)购于美国杜邦公司;商用铂碳催化剂(Pt/C,质量分数20%)购于Alfa Aesar公司;高纯氮气、氩气和氧气购于释源精业气体有限公司;去离子水为自制.
Rigaku D/Max-2500 型X 射线衍射仪(XRD),日本Rigaku 公司;Zeiss supra 55 型扫描电子显微镜(SEM),德国Carl Zeiss AG 公司;JEOL JEM-2100 型透射电子显微镜(TEM),日本电子株式会社;HD-2700型球差校正扫描透射电子显微镜(AC-HAADF-STEM),德国CEOS GmbH 公司;ESCALAB 250型X射线光电子能谱仪(XPS),美国ThermoFisher Scientific公司;ASAP-2460型全自动比表面与孔径分析仪,美国Micromeritics 公司;CHI760E 型电化学工作站,上海辰华仪器有限公司;旋转圆盘电极装置,美国PINE公司.
1.2 催化剂的制备
1.2.1 ZIF-8的制备 将Zn(NO3)2·6H2O(7.43 g)和2-甲基咪唑(8.21 g)分别溶解于100 mL甲醇中,超声10 min后,将硝酸锌溶液经由恒压滴液漏斗缓慢滴加引入2-甲基咪唑溶液中,室温搅拌24 h.获得的悬浮液,用甲醇以10000 r/min转速离心洗涤3次,于60 ℃真空干燥12 h,即得到ZIF-8.
1.2.2xFe/S@ZIF-8,S@ZIF-8 及Fe@ZIF-8 的制备 根据催化剂前驱体合成过程中Fe 原子与总金属(Fe+Zn)的摩尔比(x),尝试制备了不同Fe 含量的催化剂样品xFe/S@ZIF-8(x=1%,3%).以3%Fe 含量的Fe/S@ZIF-8 样品制备为例:将0.6 g ZIF-8 加入250 mL 三颈瓶中,然后加入120 mL 甲醇并超声使其完全分散;将0.0871 g KSCN 放入20 mL 去离子水中,将0.1099 g Fe(NO3)2·9H2O 放入20 mL 甲醇中,超声至完全溶解.将三颈瓶置于加热功率为500 W的微波反应器中,加热至60 ℃时缓慢滴加硝酸铁溶液,于60 ℃恒温反应35 min 后,缓慢滴加KSCN 溶液,在60 ℃下反应40 min.将得到的悬浮液抽滤分离,甲醇清洗3次,于60 ℃恒温真空中干燥12 h,即得到3%Fe/S@ZIF-8.
S@ZIF-8 的具体制备过程同上,微波加热时不添加Fe(NO3)2·9H2O.Fe@ZIF-8 的具体制备过程同上,微波加热时不添加KSCN.
1.2.3 Fe3C/Fe-SAS@SNC 的制备 将200 mg 3%Fe/S@ZIF-8 放入磁舟中,使用管式炉进行高温碳化.升温前通入30 min氮气排出残留在石英管中的空气,避免前驱体发生氧化.然后以5 ℃/min的升温速率加热至200 ℃并恒温保持1 h,再以同样的升温速率加热至1000 ℃,恒温保持2 h 后自然冷却到室温,即得到催化剂Fe3C/Fe-SAS@SNC.利用与3%Fe/S@ZIF-8相同的高温处理工艺,将制得的S@ZIF-8,Fe@ZIF-8 和1%Fe/S@ZIF-8 进行高温碳化处理,收集得到的产物分别标记为SNC,Fe@NC 和Fe-SAS@SNC[SAS意为单原子位点(Single-atomic site)催化剂,S原子掺杂的碳基催化剂记为SNC].
1.3 电化学性能测试
利用CHI760E电化学工作站,采用三电极体系在室温环境下进行电化学性能测试.使用饱和甘汞电极和碳棒作为参比电极(SCE)和对电极,使用玻碳电极[GCE,旋转圆盘电极(RDE)盘面积为0.196 cm2,旋转环盘电极(RRDE)盘面积为0.247 cm2]作为工作电极.测试前,将5 mg催化剂样品与950µL无水乙醇、50µL 5%Nafion溶液混合并超声处理30 min,以获得均匀分散的墨水状催化剂.玻碳电极依次用0.5,0.15和0.05µm氧化铝粉末抛光打磨并用去离子水洗涤.利用移液枪将10µL催化剂墨水数次滴加到电极(RDE负载量为0.255 mg/cm2,RRDE负载量为0.202 mg/cm2)表面,旋转干燥后即完成工作电极的制备.
负载催化剂的工作电极分别在N2气饱和与O2气饱和的0.1 mol/L KOH 和0.5 mol/L H2SO4电解液中进行循环伏安(CV)和线性扫描伏安(LSV)测试.CV扫描速率为100 mV/s,待CV曲线稳定后,在不同转速(200~2500 r/min)下以5 mV/s的扫描速率获得氧还原极化曲线.
利用RRDE 测试曲线计算电子转移数(n)和过氧化氢产率[Yield(H2O2),%],计算公式如下:

式中:IDisk(mA)是盘电流;IRing(mA)是环电流;N(0.37)是RRDE中Pt环收集效率.
电化学活性面积(Electrochemical surface Area,ECSA)通常在非法拉第效率响应的电势范围下,采用CV 法测量得到一系列不同扫描速率(1,2,3,4,5 mV/s)的曲线,并根据下面公式计算双电层电容(Cdl):

式中:j(mA/cm2)为某一扫描速率对应的电流密度;v(mV/s)为扫描速率;m(mg/cm2)为附着于电极上催化剂的负载量;CGC(0.2 F/m2)是玻碳电极的比电容.
所涉及到的电极电位均已根据能斯特方程转换为相对于可逆氢电极(RHE)电势(ERHE,V),计算公式为

2 结果与讨论
2.1 XRD表征
根据XRD 表征结果可以看出,前驱体Fe@ZIF-8,S@ZIF-8,1%Fe/S@ZIF-8 与3%Fe/S@ZIF-8 和ZIF-8的晶相一致,说明掺入Fe,N元素后的样品仍能保持原始的ZIF-8结构[图1(A)].图1(B)是相应前驱体经高温碳化后得到的Fe@SNC,SNC,Fe-SAS@SNC及Fe3C/Fe-SAS@SNC的XRD谱图.由图1(B)可知,Fe-SAS@SNC在2θ=26°处观察到强峰,其对应于石墨碳的(002)面,且除碳峰外没有观察到其它强的衍射峰.此外,Fe3C/Fe-SAS@SNC 与Fe-SAS@SNC 相比,在2θ=37.75°,39.81°,40.64°,42.89°,43.75°,44.57°,44.99°和45.87°处各有一组峰出现,其对应于Fe3C 的(210),(002),(201),(211),(102),(220),(031)和(440)晶面(JCPDS No.85-1317),表明在Fe3C/Fe-SAS@SNC 样品中存在Fe3C.Fe3C与Fe单原子的协调作用能够提高Fe-Nx位点的反应活性,有利于提升氧还原性能[20].
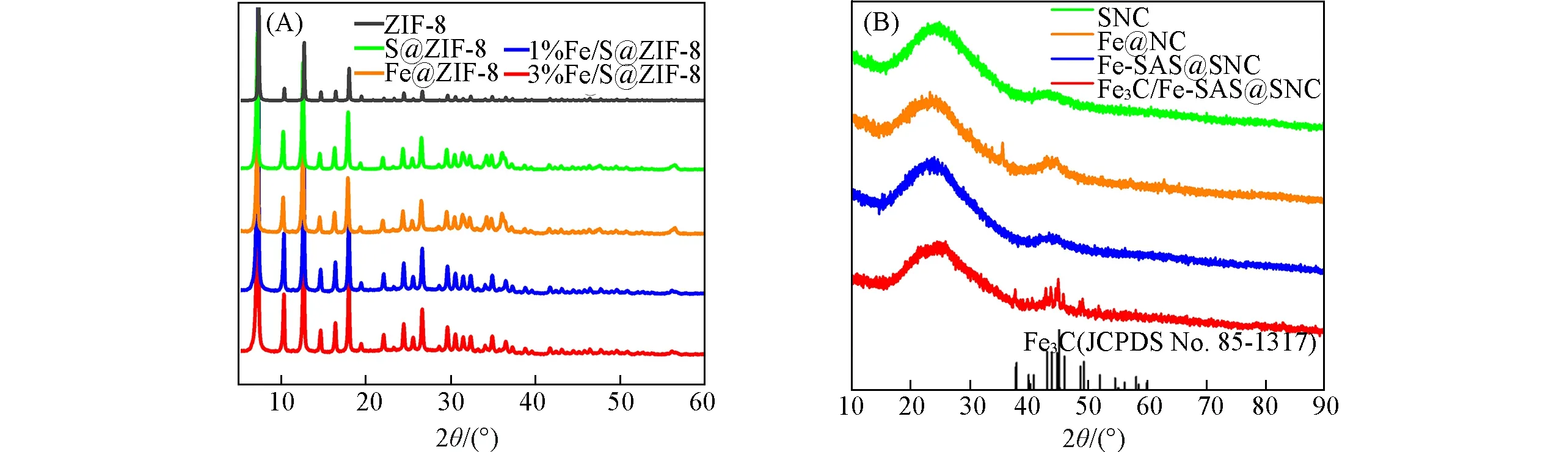
Fig.1 XRD patterns of ZIF⁃8,S@ZIF⁃8,Fe@ZIF⁃8,1%Fe/S@ZIF⁃8 and 3%Fe/S@ZIF⁃8(A),and SNC,Fe@NC,Fe⁃SAS@SNC,Fe3C/Fe⁃SAS@SNC and Fe3C(B)
2.2 SEM,TEM和HAADF-STEM表征
图2为前驱体样品及衍生材料的形貌结构图.图2(A)显示ZIF-8呈表面光滑的多面体形状,平均尺寸约150 nm.图2(B)是与Fe(NO3)3·9H2O和KSCN反应后得到的3%Fe/S@ZIF-8前驱体的形貌.可以看出,反应之后其大致保持ZIF-8 多面体结构,且表面粗糙,存在交错的纳米颗粒.由高温碳化后的SEM照片可以看到,高温碳化之后的催化剂仍保持原始的多面体形貌,表面变得更加粗糙且孔结构增多,大量碳纳米管交错生长[图2(C)和(D)].造成这一现象的主要原因是,高温碳化过程中Zn以气体形式挥发并留下大量的孔.分级多孔结构不仅有利于暴露大量的电催化活性位点,还可以促进电解质和电荷的传输.此外,金属盐的适量引入催化了大量碳纳米管的生长,也可以进一步加速电荷的传输,有助于提升ORR催化活性[25].

Fig.2 SEM images of ZIF⁃8(A),3%Fe/S@ZIF⁃8(B)and Fe3C/Fe⁃SAS@SNC(C,D)
进一步对高温碳化后的Fe3C/Fe-SAS@SNC进行TEM和HAADF-STEM表征.从图3(A)可以明显看出,催化剂表面生长一层密集的碳纳米管,局部放大后可见其呈中空结构,这有利于催化过程中电解质和电荷的传输[图3(B)].同时,如图3(C)和(D)所示,放大的Fe3C/C结构显示为Fe3C纳米颗粒被包裹在多孔碳中的核壳结构中,在Fe3C/C的核壳结构中,包裹的纳米颗粒晶格条纹间距为0.211 nm,与Fe3C 的(211)晶面吻合[图3(E)],而条纹间距0.346 nm 的晶格则对应于石墨化碳(002)晶相.Fe3C 纳米颗粒上覆盖的石墨碳层可以避免电催化过程中Fe3C的聚集,增强稳定性.同时,石墨化碳层包裹的纳米晶体Fe3C 的核壳结构可作为ORR 催化活性位点,有助于提高氧还原催化活性[26].图3(F)显示,Fe3C/Fe-SAS@SNC存在大量孤立的单原子,同时可以明显看出一些金属团簇,这与XRD的结果一致.
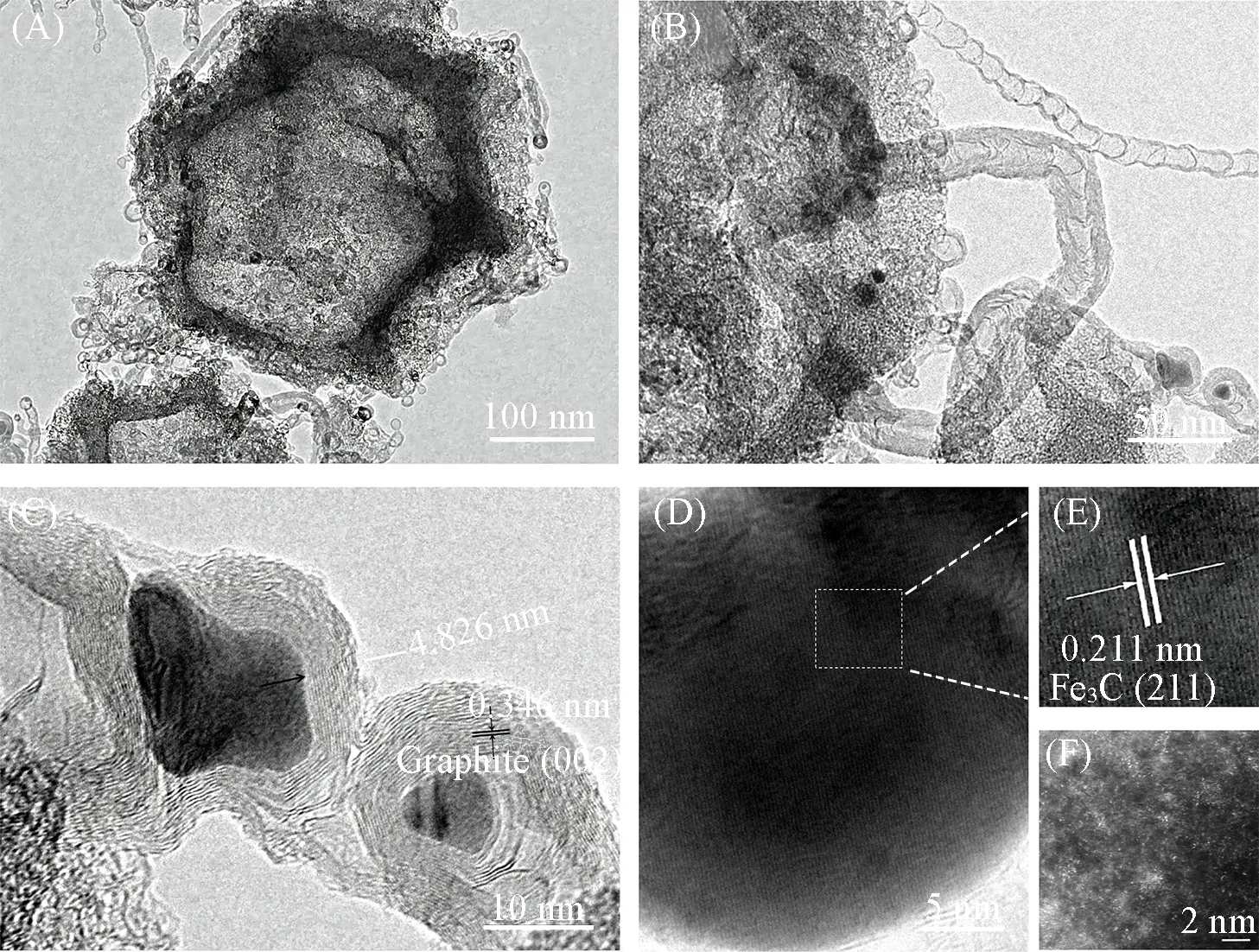
Fig.3 TEM(A―E)and HAADF⁃STEM(F)images of Fe3C/Fe⁃SAS@SNC
2.3 XPS表征
X 射线光电子能谱(XPS)是研究催化剂样品表面元素组成和电子状态的常用方法.如图4(A)和表1 所示,在Fe3C/Fe-SAS@SNC 的XPS 全谱中出现了C1s(283.81 eV),O1s(532.44 eV),Fe2p(710.08 eV),N1s(399 eV)和S2p(163.81 eV)的特征峰,证实了Fe 和S 已成功掺杂到了ZIF-8 前驱体中.另外,Fe3C/Fe-SAS@SNC 的XPS 谱中只有微弱的Fe 峰(摩尔分数为0.63%),结合TEM 图像,推测原因为,催化剂样品表面的Fe3C 纳米颗粒被石墨碳层包裹.与其它样品相比,Fe 和S 的适量引入使Fe3C/Fe-SAS@SNC 样品的N 含量提升,可提高活性位点数量,并进一步提高材料的氧还原性能.图4(B)为Fe3C/Fe-SAS@SNC 的高分辨率Fe2pXPS 谱.其中,Fe2+谱峰分别位于711.1 和722.1 eV 处,Fe3+谱峰分别位于713.4和725.6 eV处,表明样品中存在Fe-Nx活性位点,从而增加了ORR催化活性.高分辨率S2pXPS谱中的3个峰163.9,165.1和168.5 eV分别对应C=S—C键、C—S—C键和C—SOx—C键[图4(C)],其中C=S—C和C—S—C噻吩型硫结构可以通过改变碳骨架的电子结构来降低过电位,促进氧还原反应的发生[21].

Fig.4 XPS spectra of Fe3C/Fe⁃SAS@SNC,Fe⁃SAS@SNC,Fe@NC and SNC(A),and high resolution XPS spectra of Fe3C/Fe⁃SAS@SNC for Fe2p(B)and S2p(C)
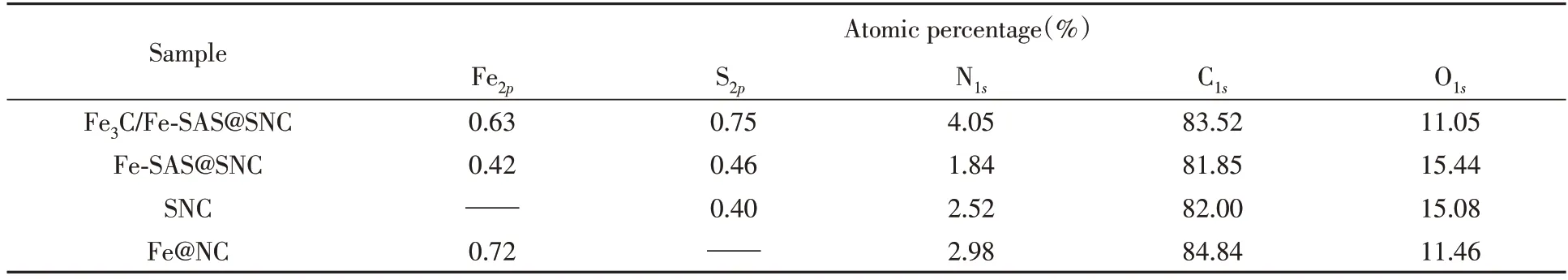
Table 1 Atomic percentage of the samples as measured by XPS
Fe3C/Fe-SAS@SNC的N1sXPS谱卷积为5个峰,分别对应吡啶N(398.5 eV)、Fe-N(399.5 eV)、吡咯N(400.7 eV)、石墨态N(401.8 eV)和氧化态N(404.7 eV)[图5(A)].根据表2 和图5(A)~(D)可知,Fe3C/Fe-SAS@SNC样品中具有相对高含量的吡啶N(1.05%)和石墨态N(1.20%),且其吡啶N和石墨态N的含量分别高于Fe-SAS@SNC(吡啶N占0.44%和石墨态N占0.34%),SNC(吡啶N占0.78%和石墨态N占0.39%)和Fe@NC(吡啶N占0.70%和石墨态N占0.66%).目前研究表明,吡啶N可以调整局部Fe-N-C活性电子结构,降低O2在相邻碳原子上的吸附能垒以促进氧还原过程,而石墨态N可加速氧还原过程中的电子转移,改善催化剂的极限电流密度[18].此外,Fe-N的存在进一步证明Fe元素已经被掺入氮掺杂的碳基体中,且Fe-N是ORR反应的有效电催化活性位点[27].因此,理论上Fe3C/Fe-SAS@SNC催化剂样品可利用的活性位点数量最多,具有比其它样品更优异的氧还原催化性能.
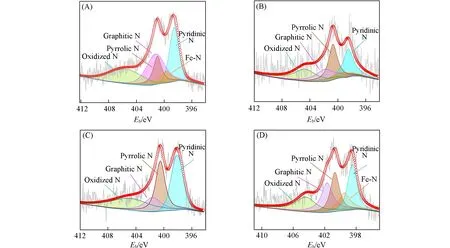
Fig.5 High resolution XPS spectra of N1s of Fe3C/Fe⁃SAS@SNC(A),Fe⁃SAS@SNC(B),SNC(C),and Fe@NC(D)
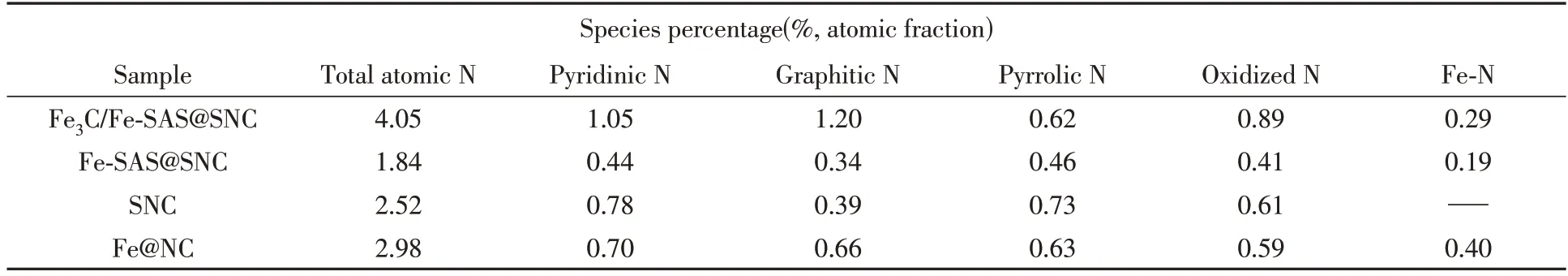
Table 2 XPS atomic percentage of different types of N of the obtained samples
2.4 氮气吸附-脱附性能
通过氮气的吸附-脱附等温曲线可以表征催化剂的比表面积和多孔性能,进一步了解催化剂的微观结构.如图6(A)和(C)所示,掺杂Fe,S样品的氮气吸附-脱附等温线都属于IV型等温线,并且在相对压力0.4~1.0范围内观察到明显的滞回曲线,表明催化剂样品中存在介孔结构,有利于暴露催化位点及促进传质[28].在p/p0>0.9处,氮气的快速吸收证明催化剂材料中存在大孔,在p/p0<0.1的低压下,较高的氮气吸收量证实了催化剂材料中微孔的存在.
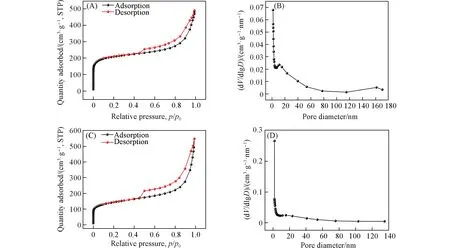
Fig.6 N2 adsorption⁃desorption isotherms(A,C) and pore size distributions(B,D) of Fe3C/Fe⁃SAS@SNC(A,B)and Fe⁃SAS@SNC(C,D)
根据表3 结果,Fe3C/Fe-SAS@SNC 的BET 比表面积为673 m2/g,大于Fe-SAS@SNC 的BET 比表面积,且具有更高的相对微孔含量.微孔的数量通常被认为是ORR催化的关键影响因素之一,微孔的存在为更多活性位点提供了负载场所,可促进ORR催化活性的提升[28].另外,从图6(B)和(D)中孔径分布曲线可以看出,大部分的孔直径在30 nm以下,表示催化剂材料中存在分级的微/介孔结构.催化剂Fe3C/Fe-SAS@SNC中较大的比表面积将会暴露出更多的催化活性位点,分层的多孔纳米结构有利于电解质的快速传输和更多催化活性位点的暴露,这些均将促进反应加速进行,提升ORR电催化性能.

Table 3 Pore characteristics of Fe3C/Fe-SAS@SNC and Fe-SAS@SNC products
2.5 氧还原性能
2.5.1 碱性条件下的ORR 性能 使用RDE 作为工作电极,与参比电极和对电极组成的三电极体系,研究所制备的催化剂的ORR电催化性能.首先,利用CV以100 mV/s的扫描速率分别在氮气和氧气饱和的0.1 mol/L KOH 溶液中对催化剂的催化活性进行测试[图7(A)].以Fe3C/Fe-SAS@SNC 为例,Fe3C/Fe-SAS@SNC 的CV 曲线在氮气饱和的电解液中未见明显的氧化还原峰,而在氧气饱和的电解质中,Fe3C/Fe-SAS@SNC 表现出了明显的还原峰(0.872 V,vs.RHE),表明催化剂样品具有ORR 催化活性.进一步利用LSV 在相同的条件下研究了Fe3C/Fe-SAS@SNC,Fe-SAS@SNC,Fe@NC 和SNC 的ORR 催化活性,并与商用20%Pt/C 进行比较.如图7(B)所示,Fe3C/Fe-SAS@SNC 的起始电位为1.02 V(vs.RHE),半波电位为0.880 V(vs.RHE),与Fe-SAS@SNC,Fe@NC 和SNC 相比,拥有更正的半波电位(E1/2)和更大的极限扩散电流密度.与其它催化剂样品相比,引入Fe,S 元素的Fe3C/Fe-SAS@SNC具有更高的氧还原催化活性.同时,催化剂本身的多孔碳纳米管结构有利于气体和电子的快速传输,从而提升了ORR催化活性.

Fig.7 CV curves of Fe3C/Fe⁃SAS@SNC in N2⁃saturated and O2⁃saturated 0.1 mol/L KOH electrolyte(A),ORR polarization curves in O2⁃saturated 0.1 mol/L KOH electrolyte(B)
通过计算处理得到了所有催化剂样品塔菲尔曲线线性极化区的斜率,塔菲尔斜率越小,其反应动力学越快,相应的ORR 电催化活性越好.如图8(A)所示,Fe3C/Fe-SAS@SNC 催化剂的塔菲尔斜率为94 mV/dec,低于Fe-SAS@SNC,Fe@NC和SNC,最接近商业Pt/C的塔菲尔斜率,说明Fe3C/Fe-SAS@SNC催化剂对ORR反应表现出良好的动力学过程.此外,ECSA是表征材料能发生催化作用的面积,可以通过不同扫描速率下的CV曲线获得.如图8(B)和(C)所示,经测试计算得出,Fe3C/Fe-SAS@SNC催化剂的ECSA为535 m2/g,高于Fe-SAS@SNC(475 m2/g),SNC(415 m2/g)和Fe@NC(140 m2/g),也表明掺杂杂原子策略可以对催化剂的孔结构进行有效调节,从而调节催化活性位点面积,调控Fe,S引入量后得到的Fe3C/Fe-SAS@SNC催化剂具有更优的ORR催化性能.
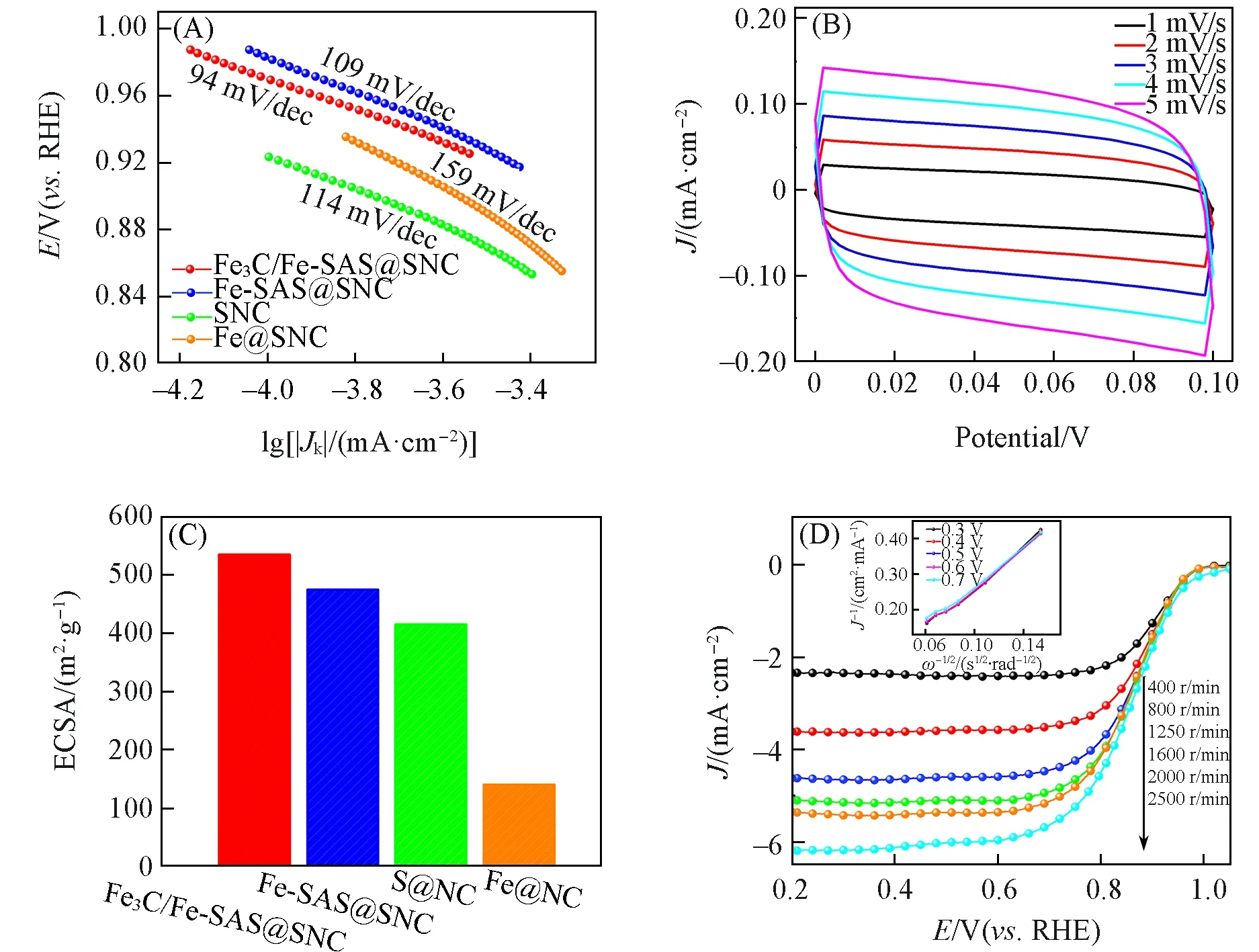
Fig.8 Tafel slope of different catalysts(A),CV curves at different scan rates of Fe3C/Fe⁃SAS@SNC(B),the corresponding ECSA for ORR of different catalysts(C),LSV curves at different rotating speeds of Fe3C/Fe⁃SAS@SNC(inset:K⁃L plots at different potentials)(D)
通过测试Fe3C/Fe-SAS@SNC 在400,800,1250,1600,2000 和2500 r/min 不同转速下的氧还原线性极化曲线,并拟合不同电极电位下相应的Koutecky-Levich(K⁃L)曲线[图8(D)插图],来研究Fe3C/Fe-SAS@SNC催化剂催化氧还原的动力学过程.从图8(D)可以看出,催化剂极限电流随转速的增大而增大,说明氧还原过程受氧扩散速率的影响.同时,Fe3C/Fe-SAS@SNC 催化剂的K⁃L曲线中J−1与ω−1/2显示出良好的线性关系,证明其催化的氧还原反应属于一级动力学反应.
直接四电子过程是氧还原的理想反应途径,但实际条件下往往伴随二电子过程副反应并产生H2O2,会降低能量转换效率并破坏电极[10].因此,利用旋转环盘电极装置对Fe3C/Fe-SAS@SNC 催化剂进行了测试,分别计算了其转移电子数和H2O2产率,确定催化剂对四电子还原过程的选择性.如图9(A)所示,Fe3C/Fe-SAS@SNC 的电子转移数在3.86~4 的范围内,比商业Pt/C 更加稳定.此外,Fe3C/Fe-SAS@SNC在0.2~0.8 V的电位范围内具有非常低的H2O2产率(低于7%),明显低于商业Pt/C的H2O2产率[图9(B)],这些结果证实了Fe3C/Fe-SAS@SNC催化剂对四电子途径具有高选择性,其催化的氧还原过程主要经历四电子途径.
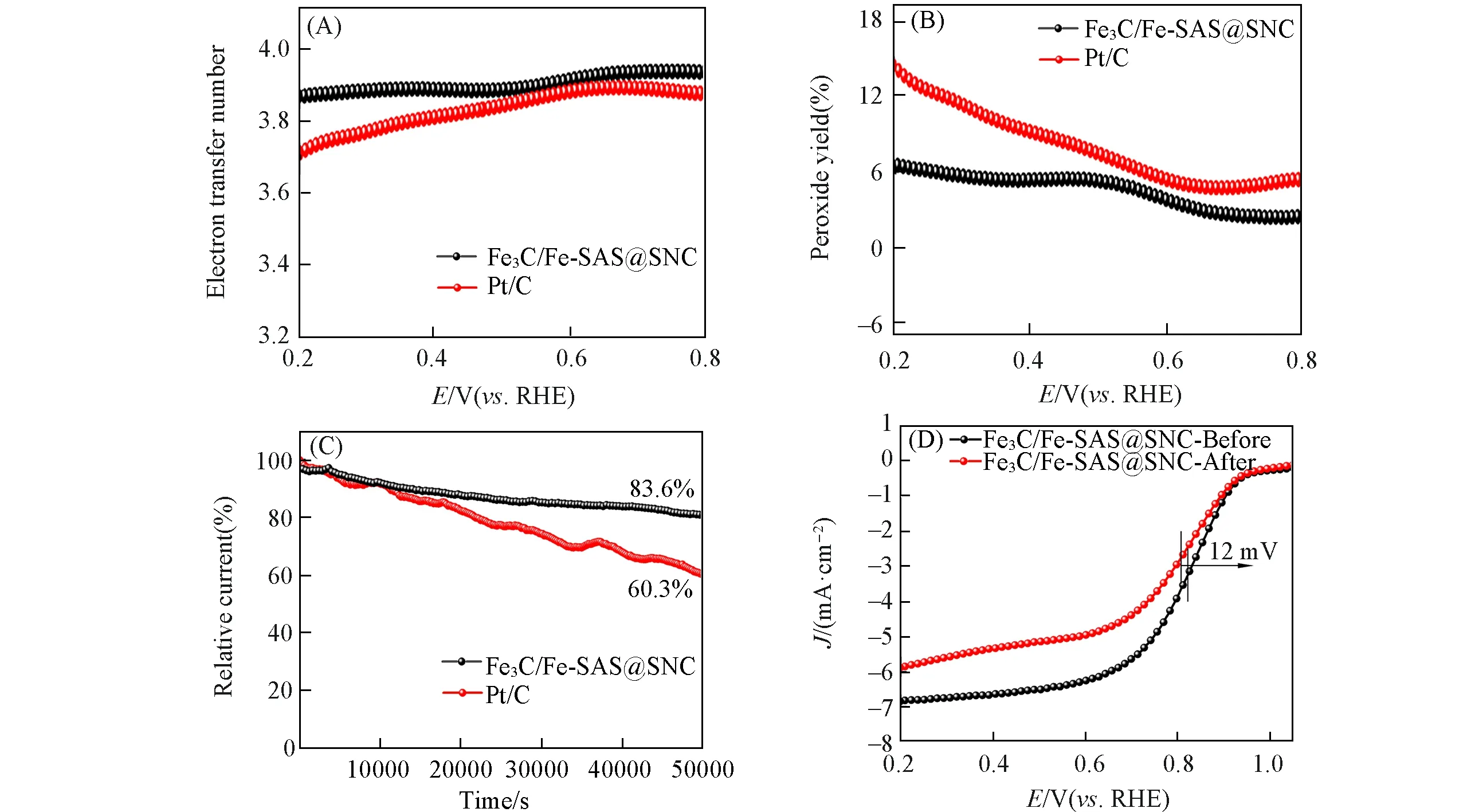
Fig.9 ORR catalytic performance in O2⁃saturated 0.1 mol/L KOH electrolyte
除了高的ORR电催化活性,良好的稳定性也是商业应用的优异催化剂必不可少的特征.因此,在1600 r/min 的O2气饱和0.1 mol/L KOH 溶液中,对Fe3C/Fe-SAS@SNC 进行50000 s 计时电流测试(i⁃t).图9(C)是Fe3C/Fe-SAS@SNC 和Pt/C 在0.5 V 电位下测试得到的i⁃t曲线.可以看出,Fe3C/Fe-SAS@SNC在连续工作50000 s后仍然可以保持初始电流的83.6%,明显高于商业Pt/C的保持率(60.3%).图9(D)是Fe3C/Fe-SAS@SNC在i-t测试前后在1600 r/min下的LSV曲线,可以看出,Fe3C/Fe-SAS@SNC的LSV曲线趋势基本保持不变,E1/2仅负移了12 mV.因此,Fe3C/Fe-SAS@SNC催化剂在0.1 mol/L KOH中具有优于商业Pt/C的稳定性.
2.5.2 酸性条件下的ORR 性能 在O2饱和的0.5 mol/L H2SO4电解液中,利用同样的三电极体系对Fe3C/Fe-SAS@SNC,Fe-SAS@SNC,Fe@NC和SNC催化剂材料进行旋转圆盘测试,研究其ORR电催化性能.结果显示,Fe3C/Fe-SAS@SNC 催化剂材料也具有最优的氧还原催化性能,和碱性电解液环境下得出的结论一致.图10(A)的LSV 曲线显示,在所有催化剂中,Fe3C/Fe-SAS@SNC 对ORR 过程表现出最高的催化活性,其起始电位为0.892 V(vs.RHE),半波电位达到0.785 V(vs.RHE).进一步从塔菲尔曲线拟合得到Fe3C/Fe-SAS@SNC 催化剂的塔菲尔斜率为94 mV/dec,低于Fe-SAS@SNC,Fe@NC 和SNC,最接近商业Pt/C,相应的ORR电催化活性最好[图10(B)].
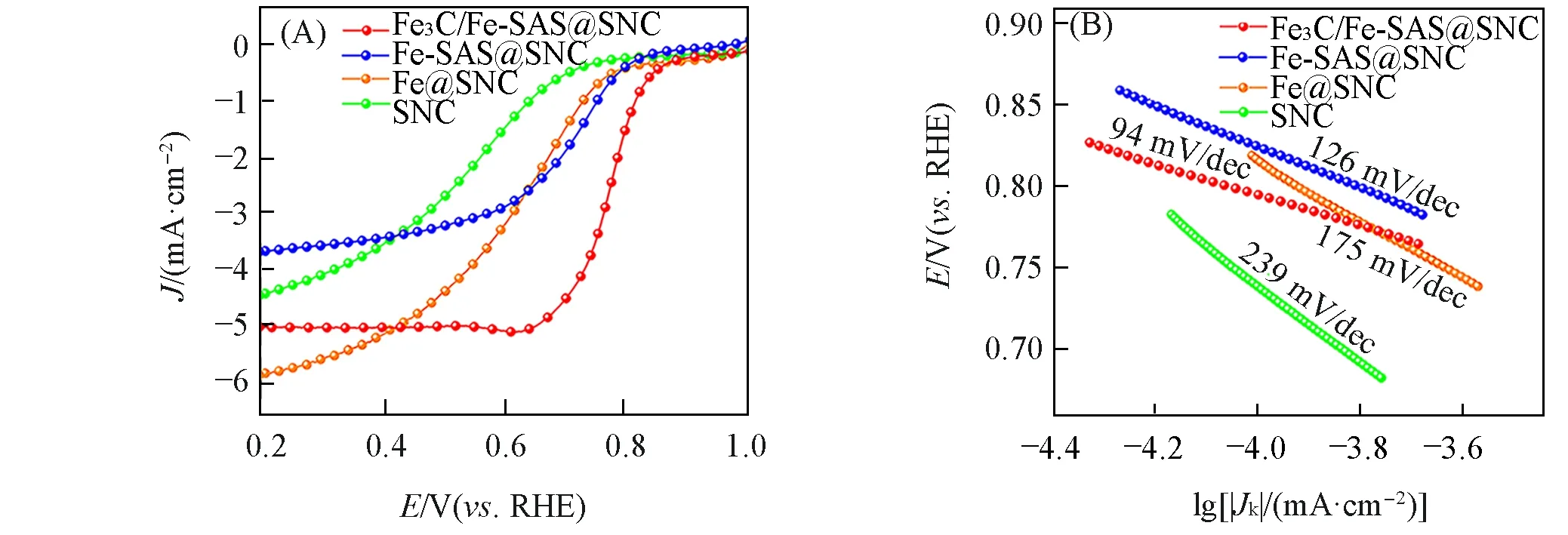
Fig.10 ORR polarization curves(A) and Tafel slope(B) of Fe3C/Fe⁃SAS@SNC,Fe⁃SAS@SNC,Fe@NC,and SNC in O2⁃saturated 0.5 mol/L H2SO4 electrolyte
测试了Fe3C/Fe-SAS@SNC 在不同转速下的氧还原线性极化曲线,并拟合了不同电极电位下其Koutecky-Levich(K-L)曲线[图11(A)及插图].可见,催化剂极限电流随转速的增大而增大,说明氧还原过程受氧扩散速率的影响.同时,酸性环境下Fe3C/Fe-SAS@SNC催化剂的K-L曲线中J−1与ω−1/2显示出良好的线性关系,进一步证明其催化的氧还原反应属于一级动力学反应.利用旋转环盘电极装置在酸性电解质中测试了Fe3C/Fe-SAS@SNC 催化剂的转移电子数和H2O2产率.Fe3C/Fe-SAS@SNC 的电子转移数在3.94~4 的范围内,同商业Pt/C 相比更加稳定[图11(B)].此外,Fe3C/Fe-SAS@SNC 在0.2~0.8 V电位范围内具有非常低的H2O2产率(低于3.5%),明显低于商业Pt/C的H2O2产率[11(D)],这些结果均证实该催化剂对四电子过程具有高选择性,进一步说明了杂原子掺入后的Fe3C/Fe-SAS@SNC材料在酸性电解液中也具有良好的氧还原催化活性.为了探究催化剂在酸性介质中的稳定性,在1600 r/min 的O2气饱和的0.5 mol/L H2SO4溶液中对Fe3C/Fe-SAS@SNC 进行计时电流测试.图11(D)是Fe3C/Fe-SAS@SNC和Pt/C在0.5 V的电位下测试得到的i⁃t曲线.可以看出,Fe3C/Fe-SAS@SNC在连续工作50000 s后仍然可以保持初始电流的80.9%,而商业Pt/C 在连续工作30000 s 后仅保持初始电流的45%.图12 是Fe3C/Fe-SAS@SNC 在i⁃t测试前后1600 r/min 下的LSV曲线.可以看出,Fe3C/Fe-SAS@SNC的LSV曲线趋势基本保持不变,E1/2仅负移了15 mV.因此,Fe3C/Fe-SAS@SNC 催化剂在酸性电解液中也具有良好的稳定性.
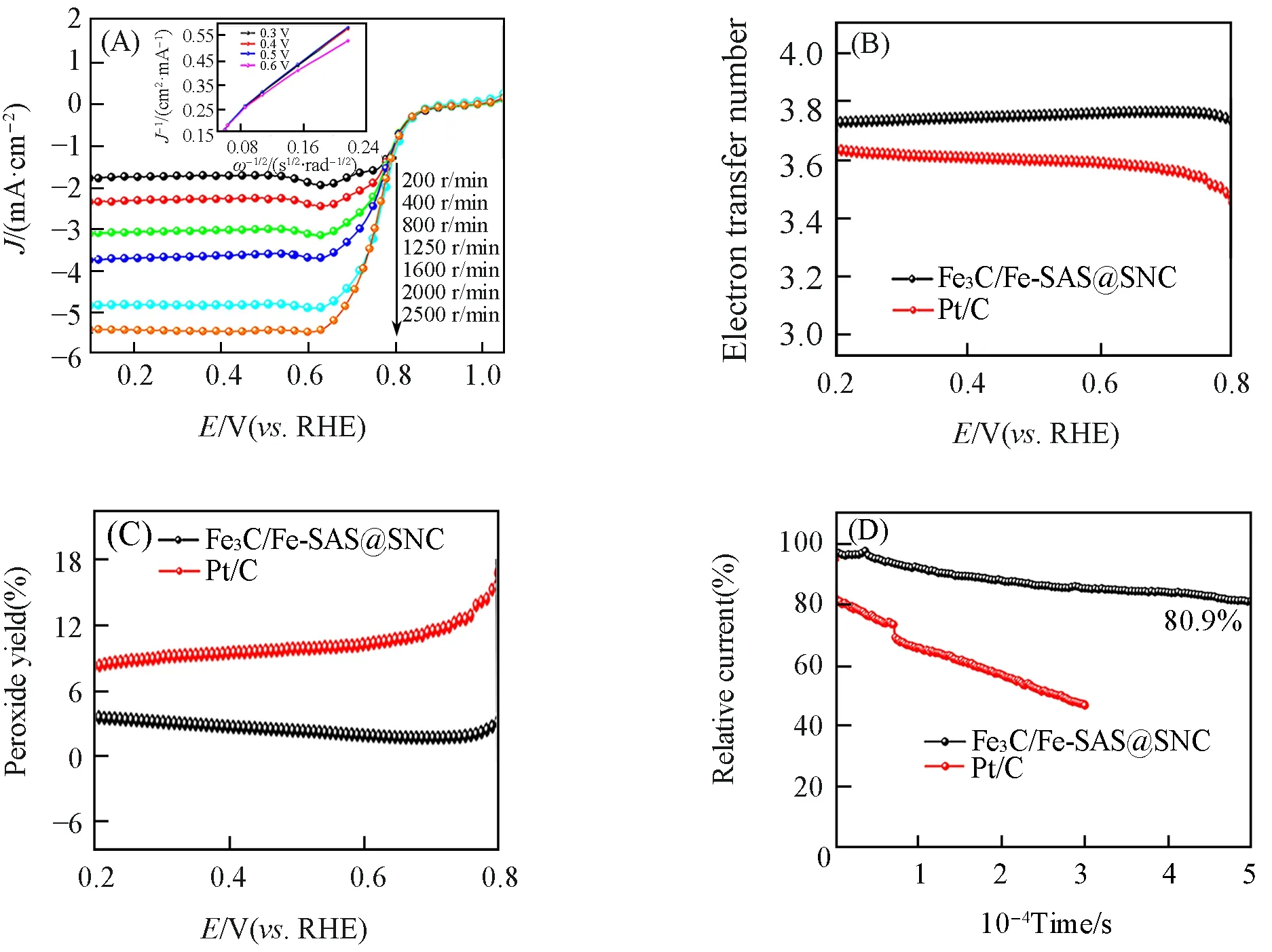
Fig.11 LSV curves of Fe3C/Fe⁃SAS@SNC at different rotating speeds(inset: K⁃L plots at diffe⁃rent potentials)(A),electron transfer number(B),H2O2 yield plots(C),chronoamperomet⁃ric(i⁃t) responses of Fe3C/Fe⁃SAS@SNC and 20%Pt/C in O2⁃saturated 0.5 mol/L H2SO4 electrolyte(D)
另外,由表4 中数据可见,Fe3C/Fe-SAS@SNC催化剂与同类催化材料相比也具有相对突出的ORR催化活性[29~37].
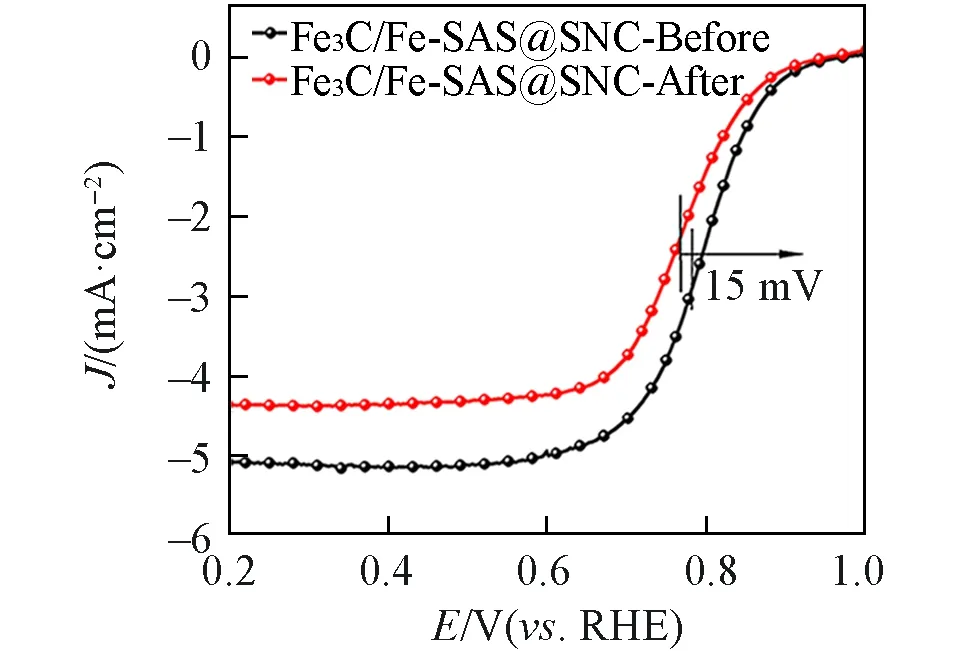
Fig.12 ORR polarization curves of Fe3C/Fe⁃SAS@SNC before and after Chronoamperometric(i⁃t) test(1600 r/min)in O2⁃saturated 0.5 mol/L H2SO4 electrolyte
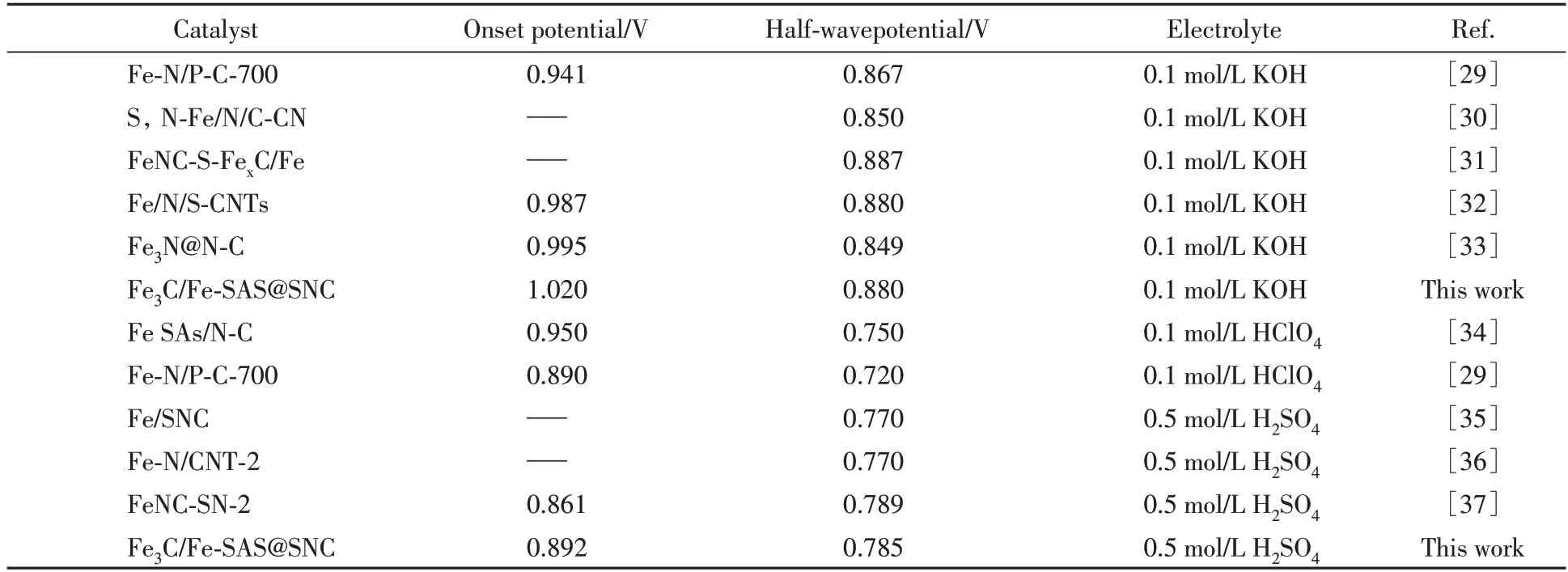
Table 4 Fe-based electrode materials for ORR
3 结 论
采用微波加热和高温碳化技术,以ZIF-8 为前驱体,通过在甲醇-水双溶剂体系中先后引入Fe(NO3)3·9H2O和KSCN,制备了具有高比表面积和高孔隙率的Fe3C/Fe-SAS@SNC氧还原催化剂,并利用XRD,SEM,TEM,XPS 及BET 等表征手段进行了分析.结果表明,高温热解过程中低沸点的Zn 挥发使Fe3C/Fe-SAS@SNC 催化剂拥有明显的分级微/介孔结构,比表面积达到673 m2/g,且表面大量碳纳米管交错生长,有利于加快电解质传输、暴露更多催化活性位点.适量Fe的引入可以在催化剂表面形成石墨化碳层包裹的Fe3C纳米颗粒,作为ORR催化活性位点,进而提升催化剂氧还原催化活性.适量S的掺入不仅使催化剂中吡啶N和石墨态N等ORR活性点含量增多,还可以形成类噻吩硫结构以促进氧还原反应的发生.表明杂原子的合理掺入使Fe3C/Fe-SAS@SNC催化剂在碱性和酸性条件下均表现出优异的ORR 催化性能.本文开发的简单可行、低成本、高催化活性和高稳定性的非贵金属催化剂Fe3C/Fe-SAS@SNC 的制备方法,为非贵金属催化剂的发展提供了新思路.但是,高温煅烧的不可控性导致对此类催化剂活性位点的认识不足,未来可深入研究其作用机理,以进一步指导催化剂的合成.
猜你喜欢
昆钢科技(2022年1期)2022-04-19
汽车工程师(2021年12期)2022-01-18
食品安全导刊(2021年20期)2021-08-30
纺织科学研究(2021年7期)2021-08-14
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
商品与质量(2019年42期)2020-01-17
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
