
XAFS技术在单原子电催化中的应用
2022-09-19 06:28汪思聪庞贝贝刘潇康
高等学校化学学报 2022年9期
汪思聪,庞贝贝,刘潇康,丁 韬,姚 涛
(中国科学技术大学国家同步辐射实验室,合肥 230029)
单原子催化剂因其独特的原子结构和电子性质、极高的原子利用率以及在多种化学反应中优异的整体性能,被广泛应用于多种涉及能源转换与储存的电化学反应中[1~5].然而,由于常规表征方法的局限,其活性位的明确及原子电子结构的探测一直是一大难题.X射线吸收精细结构谱(X-Ray absorption fine structure spectroscopy,XAFS)具有对中心吸收原子的局域结构和化学环境敏感的特征,能够在原子尺度上给出吸收原子周围几个邻近配位壳层的结构信息,包括配位原子的种类及其与中心原子的距离、配位数和无序度等[6~10].这些独特的性质使得XAFS技术成为最适合探测单原子催化剂活性中心原子局域结构的手段之一.另外,由于XAFS 方法对样品形态要求不高,可测样品包括晶体、粉末、薄膜、液体等,且不会对样品产生明显的破坏[11],该技术对电化学原位测试的兼容性极高,能很好地探测单原子催化剂活性位点在电化学反应过程中动态结构变化,为单原子电催化剂的作用机理提供关键证据.因此,近年来,XAFS技术被广泛应用于单原子电催化剂的设计与表征中,并逐渐成为单原子电催化剂最常用的结构表征手段之一.
1 XAFS技术介绍
X射线穿过厚度为d的样品后,其强度I0会因为样品的吸收而衰减为IT.基于朗伯比尔定律,可以定义样品的X射线吸收系数(µ):

吸收系数随入射X 射线光子能量E变化的曲线就是X 射线吸收谱(X-Ray absorption spectroscopy,XAS).由于电子跃迁的选择性,XAS谱会在一些高度元素敏感的位置产生特征起跳,吸收系数发生突变,称为吸收边.早在1920年,Friche和Hertz即发现硬X射线能量区XAS谱的部分吸收边后会出现一系列微小的摆动和振荡,并敏锐地意识到这些振荡的规律性特征,将其称之为X射线吸收的精细结构(XAFS)[12,13].但直到20世纪80年代,XAFS现象才得到了比较完整可信的解释,相关的实验技术也得以迅猛发展[14].20 世纪90 年代至今,伴随着第二代/第三代同步辐射光源的普及,XAFS 技术日趋成熟,并发展出快速扫描XAFS(QXAFS)[15]、能量色散XAFS(DXAFS)[16]及泵浦探测XAFS(pump-probe XAFS)[17]等多种新型实验技术,在材料科学领域获得了广泛应用.
1.1 XAFS的原理和特征
XAFS现象本质上来源于入射X射线激发出的光电子与周围原子的散射作用.对绝大部分元素而言,XAFS谱可以粗略地分为两部分[18]:X射线吸收近边结构(X-Ray absorption near edge structure,XANES)和扩展X 射线吸收精细结构(Extended X-ray absorption fine structure,EXAFS)(图1).两个区域的XAFS 谱来源于不同种类的X 射线散射作用,对不同的结构信息敏感.
XANES 一般指吸收边前10 eV 至吸收边后30~50 eV范围的XAFS结构.在这一区域中,由于入射X 射线能量与吸收边能量接近,产生的光电子动能较低,因而受近邻原子的电位和光电子本身的多重散射影响很大[18].XANES对费米能级附近的电子态敏感,能提供包括中心原子价态、近邻配位种类、材料晶体结构等信息在内的多种材料结构信息[19].
EXAFS一般指吸收边后50~1000 eV范围的XAFS结构.在这一区域中,入射X射线能量远高于吸收边能量,产生的光电子动能较高,受近邻原子的电子轨道影响很小,起主导作用的是激发出的光电子与周围原子之间的单重散射效应[18].经过近邻壳层原子单散射之后的光电子与出射波相干涉,引起振幅不大的振荡,形成了EXAFS结构.EXAFS与晶体结构等长程有序结构无关,但对原子的近邻空间构型非常敏感.
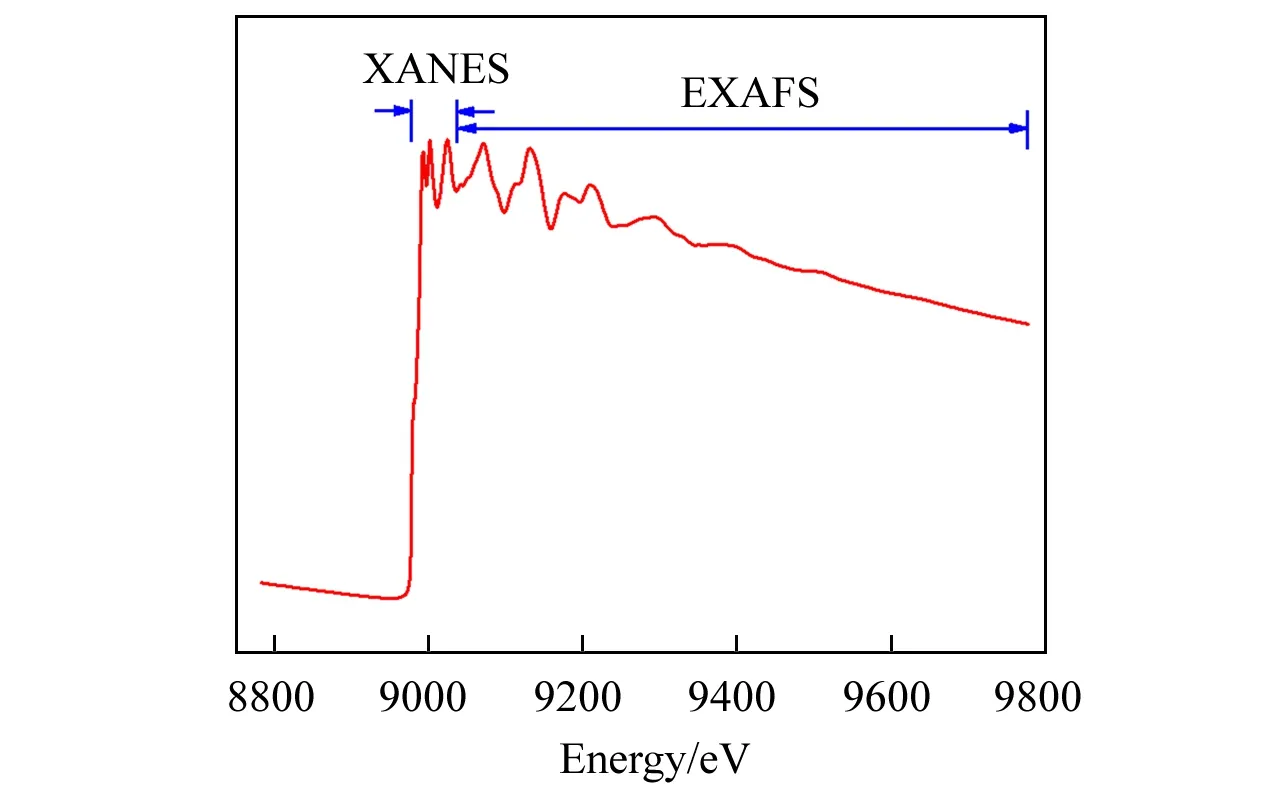
Fig.1 Cu K⁃edge XAFS spectrum for Cu foil
1.2 XAFS的解析
XAFS 的解释经过近百年多位科学家的发展完善现在已趋于成熟,其精确的数学模型已得到构建.但由于其数学表述的复杂性,目前XAFS 的解析依赖经验规律和模拟计算的结合,需要大量的实例分析经验和一定的计算化学功底.由于篇幅有限,在这里仅做简单的介绍.
XANES 的来源相当复杂,部分曲线特征可能是多个结构信息的叠加,因此难以进行定量精确分析.由XANES 曲线可以得到的最直观的信息就是待测样品中目标元素的平均价态.对于大部分金属元素而言,随着价态的升高,XAS吸收边位置会整体向高能量方向移动[20,21].因而通过样品的XANES曲线与已知标样的XANES曲线的吸收边位置对比,即可判断样品的大致价态.对Pt金属的L边而言,其XANES曲线在吸收边后有一个明显的特征峰,称之为“白线峰”.白线峰的面积与金属价态相关,金属价态越高,白线峰面积越大[22,23].因而对Pt而言,样品目标元素的价态可以借助白线峰面积的比较进行一定程度的粗略计算.此外,对于同一种元素而言,不同的配位结构如MN4、MO8等会导致完全不同的XANES整体形状,通过直观的形状比对可以判断探测元素的近邻配位种类.近年来,借助全路径多重散射(FMS)[24]、自洽场和完全相对论计算[25]等多种理论计算模型,人们可以通过FitIt,MXAN 和FDMNES 等软件工具[19]从头计算一些结构的XANES 谱[20],从而实现一定程度的XANES 谱定量分析.但总体而言,目前XANES的解析还很依赖非定量的特征比对,未来仍有广阔的发展优化空间.
相比于XANES,EXAFS主要由X射线激发出的光电子与周围原子之间的单重散射效应产生,受多重散射影响很小,物理模型较简单,定量分析也更容易.借助单散射理论,可以得出通用的标准EXAFS函数[26]:
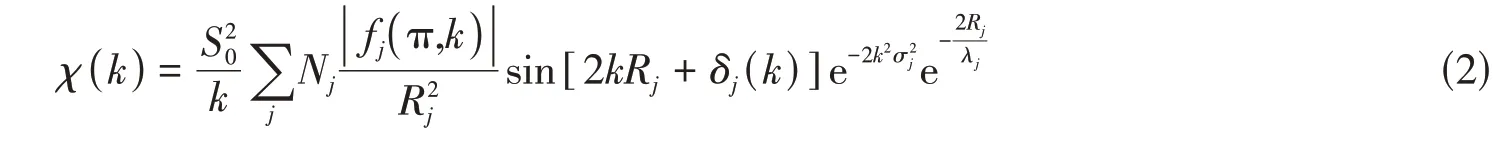
式中:χ(k)为光电子波矢k的函数.以此公式为基础直接进行EXAFS 解析仍然十分困难.但近年来,一些分析软件如Artemis等被发展出来.这些软件将EXAFS的关键影响因素:吸收系数、键长、配位数和无序度等信息抽离出来,基于非线性最小二乘法规则对EXAFS曲线进行拟合,可以得到较精确的结构信息.利用Artemis软件拟合得到的键长可以精确到0.001 nm,配位数可以精确到0.1.
1.3 XAFS实验技术
一般来说,XAFS信号的强度在总吸收强度的10%以下,因而对实验测量的精度有很高的要求.目前,主流的XAFS实验通常采用高通量同步辐射光作为光源,通过双晶单色器来获得高能量分辨的单色光,再用高性能探测器对相关信号进行探测,以得到高信噪比的XAFS 谱.为了满足不同的研究需要,已发展了多种XAFS测量方法,其中最常用的就是透射测量法和荧光测量法(图2)[19].
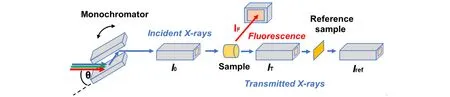
Fig.2 Schematic experimental set ups for XAFS measurement[19]
1.3.1 透射测量法 透射测量法是最直接且简便常用的收集XAFS信号的方法[6,11].将一束X射线单色光穿过样品,通过前、后电离室分别测量入射光强度(I0)和出射光强度(IT),由式(1)吸收系数的定义即可直接计算得到吸收系数.通过转动单色器晶体而改变入射光的能量进而实现X射线的能量扫描,同时同步地测量每一个能量点的I0和IT,即可得到µ(E)曲线.
透射测量法适合测量高浓度(待测元素质量百分比大于5%)的样品.在测试前,根据待测元素的吸收边能量的不同,气体电离室需要通入不同比例的气体,一般确保前后电离室对X射线的吸收均在20%左右,从而保证两个电离室都工作在线性区.此外,为提高采集信号的信噪比,对用于测试的样品有一定的要求.样品必须厚度均匀,没有透光的孔洞,颗粒尺寸不能太大,且应计算样品的厚度d以使吸收边的跳高Δµ≈1,防止后电离室信号过强或过弱.
透射测量法测试效率高,操作便捷,分析处理方便.但透射法对样品均匀性和待测元素含量有严格的要求,难以对单原子催化剂等低目标元素浓度的材料进行探测.
1.3.2 荧光测量法 荧光测量法[6,11]是另一种常用的XAFS 信号收集方法.由于X 射线的荧光产额正比于吸收系数,利用荧光探测器收集样品产生的荧光信号IF,即可获取吸收系数.为了尽可能减少弹性散射信号等本底辐射的干扰,荧光探测器通常布置在与入射光成直角的位置上,而样品则放置在与入射光和探测器均成45°角的位置上(图2).荧光测量法最常用的探测器是Lytle 探测器.在传统气体电离室探测器的前方,另加由原子序数比待测原子序数小1的元素组成的滤波片以吸收待测元素以外的其余元素产生的荧光信号,并设置索拉狭缝以隔离垂直入射光方向上复杂的非弹性散射信号,即可构建简单通用的Lytle探测器结构,因而只有放置在索拉狭缝焦点处的样品中待测元素的荧光信号才可通过由原子序数为Z−1的元素组成的滤波片和狭缝到达探测器.Lytle探测器可以探测待测元素质量百分比低至千分之一的样品的XAFS信号,足以满足绝大多数单原子催化剂的探测需求.
当待测元素的质量浓度低至十万分之一甚至百万分之一时,Lytle探测器就很难收集到高信噪比的XAFS信号了.这时就需要采用固体探测器.目前主流的固体探测器为多元高纯Ge固体探测器,其大致构筑方法是以高纯Ge 为本征半导体,分别用掺杂和离子注入形成n 区和p 区,构筑一个p-i-n 节.X射线从p区进入探测器后,在i区发生电离,产生电子-空穴对,它们在外加电场的作用下分别向正、负极移动,形成电信号.将此电信号经放大电路放大,即可获取XAFS信号.固体探测器具有极强的探测能力和极高的能量分辨率,适合探测化学环境复杂、质量分数极低的待测元素.
荧光探测法是目前单原子催化剂XAFS表征最常用的方法,其对低元素浓度样品的探测能力和对样品物理状态的高度兼容性与现阶段大部分单原子催化剂的实际情况契合.由于实际样品成分复杂,其荧光信号组成也较复杂,合理地优化光路、过滤不相关的荧光信号,尤其是针对金属含量较高的高本底样品进行相关设置,对于高质量的荧光XAFS谱的采集和后续的数据处理至关重要且具有一定的挑战性.
1.3.3 全电子产额法 全电子产额(Total electron yield,TEY)法[27]是21 世纪初开始普及的一种新式XAFS 测试方法.该方法借助真空电离室,直接在反射光方向测量样品在入射X光激发下产生的光电流,从而反推XAFS谱(图3)[28].虽然全电子产额法测量获取的信号来源很杂,包括弹性电子、非弹性电子、俄歇电子等多种光电子,但由于俄歇电子的数量远超其它来源的电子,此方法测得的实际信号不需要经过复杂的数学转化就可以获取相当精确的XAFS信号.

Fig.3 Schematic layout of the ion chamber in a total electron detection mode[28]
全电子产额法能够完全屏蔽样品“自吸收”效应的影响,信噪比优于传统的荧光法.另外,由于俄歇电子在材料中的平均自由程很小,只有1~5 nm,全电子产额法对样品的表面结构变化相当敏感,非常适合探测薄膜材料的结构和在单晶表面进行的模型催化反应.但是,全电子产额法要求待测材料与电离室电极有欧姆接触,因而对材料的导电性有较高的要求,且无法探测材料的体相结构.同时,俄歇电子产额随着原子序数的增加而逐渐减小,使得该方法更适用于轻元素吸收谱的研究.这些都使得全电子产额法只适用于特定的材料体系.
1.3.4 原位XAFS 技术 由于XAFS 对目标元素短程有序信息高度敏感,且不会对样品产生明显的破坏,该技术非常适合探测目标元素在工况条件下近邻配位结构的变化,从而为复杂电化学反应的机理研究提供关键信息.因此,近年来,各类原位XAFS(in situXAFS或operandoXAFS)技术得到了广泛的关注[19].图4展示了两种最为常用的电化学原位XAFS测试标准装置.两个装置的基本构造类似,都是将电催化剂滴涂在碳纸上作为工作电极,用Kapton胶带或Si3N4膜封住X射线入射窗口以防止电解液的泄露,再搭建经典三电极工作系统,并外接一个蠕动泵以保证反应过程中电解液的流动[19,29].其中,图4(A)中的电化学原位反应池容积较大,可以更真实地模拟实际电化学测试条件,且气泡效应不明显,工作更稳定,但由于水层较厚,而水对X射线有明显的吸收效应,因此该装置只能进行荧光模式的原位XAFS测试.而图4(B)中的电化学原位反应池则设计成扁平的圆盘状,有效降低了水层吸收的影响,既可以进行荧光模式测试,也可以进行透射模式测试,但这种设计导致了反应池容积受限,一些需要额外通入气体的电化学反应(如二氧化碳还原等)可能没有足够的空间外接气路,且气泡效应更为明显,工作状况不如图4(A)中的稳定.在实际测试中,需要根据测试需求灵活选择测试装置或自行设计定制电化学原位反应池.

Fig.4 Two typical in situ electrochemical cells[19]
在实际电化学反应中,由于化学反应的动力学过程和催化剂的结构变化往往发生在极快的时间尺度下(ns~ms 量级),发展时间分辨XAFS(Time-resolved X-ray absorption fine structure spectroscopy,tr-XAFS)技术是原位XAFS 实验中最重要的课题之一.快速扫描XAFS(Quick-scanning X-ray absorption fine structure spectroscopy,QXAFS)方法[15]是最基础的时间分辨XAFS技术.通过应用连续扫描单色器和优化的快速数据采集程序,可以在单色器快速转动的同时进行数据采集,进而将XAFS的时间分辨尺度从分钟量级压缩到百ms 量级,实现一些较缓慢的催化剂近邻配位结构变化的连续观测.例如,Zhang 等[30]利用QXAFS 技术观测到Sn 单原子稳定的Cu2O 纳米片在连续120 min 的电化学CO2RR 反应中Cu2O 逐渐还原为Cu 的过程,并结合主元成分分析(PCA)验证了Sn 单原子对Cu2O 纳米片的稳定作用;Ishiguro等[31]利用QXAFS技术研究了多种已知Pt基催化剂在电化学ORR反应中的一些独特的结构变化.另外,由于QXAFS技术本质上是对传统XAFS技术的优化,采样原理也与传统XAFS技术相同,该技术在一些场合也被用于提高传统XAFS测试的采样效率.目前,北京同步辐射装置(BSRF)、上海同步辐射光源(SSRF)和台湾光子源(TPS)均能进行QXAFS实验.
能量色散XAFS(Energy dispersive X-ray absorption fine structure spectroscopy,ED-XAFS 或DXAFS)[16,19]是一种新颖的XAFS采样技术,因其极高的时间分辨应用前景得到广泛的关注.DXAFS技术用弯晶单色器取代传统XAFS 技术中的双晶单色器,将X射线聚焦在样品处,在经过样品后依据不同的布拉格角再次发散,进而将能量信息转化为位置信息,再用二维面探测器进行信号采集(图5),一次性记录下位置和对应强度信息,即可在一次采谱中得到整个吸收谱.基于上述原理,理论上数据采集时间取决于X 射线光子入射后探测器的响应时间,因此该技术的时间分辨率远高于传统XAFS 和QXAFS,可达到µs 数量级.但目前DXAFS技术无法进行荧光模式实验,且由于样品处聚焦光斑通常为微米级大小,对样品均匀度、待测元素浓度等参数的要求较高.
泵浦-探测XAFS(Pump-probe XAFS)技术[17]是目前最先进实用的原位时间分辨XAFS 技术.泵浦-探测XAFS技术的具体实验方案多种多样,相当复杂,但本质上都是通过额外引入一束用于激发样品的激光(泵浦光),调节泵浦光与探测用X射线脉冲(探测光)的时间延迟进行多次单点探测,以获取目标材料在反应中不同时间尺度下活性位点电子结构的变化信息.泵浦-探测XAFS技术的时间分辨尺度可达ps量级[32].但由于所有的泵浦-探测XAFS实验都涉及多次单点探测,此技术对材料在实际反应条件下的稳定性、均匀性和可逆性均有相当高的要求,目前基本只适用于均相催化剂和模型催化剂,如何将此技术拓展到实际催化体系中更常见的多相催化剂仍是一大挑战.另外,受限于实验用探测器的探测能力,目前主流的pump-probe XAFS 探测实验均对样品目标元素最低浓度有一定的要求,且只能采集XANES谱.
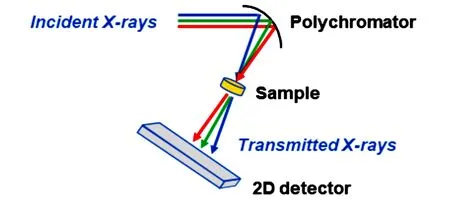
Fig.5 Schematics of a common setup for energy⁃dispersive XAFS experiment [19]
2 XAFS技术在单原子电催化中的应用
由于XAFS技术具有对目标原子短程有序信息敏感的特性,且具备探测低浓度元素相关信息的能力,该技术与有效成分浓度低、活性中心原子近邻配位结构不明确、样品状态往往呈非晶粉末态的单原子催化剂材料适配度极高.另外,XAFS相关原位实验的丰富性也使它能方便地探测电化学工况条件下的催化剂活性位点的结构变化,为探索催化剂在电化学反应中的实际作用提供关键信息.因此,近年来,XAFS技术被广泛应用于单原子的HER,OER,ORR和CO2RR等多种电化学反应中,逐渐成为单原子电催化剂不可或缺的表征手段之一.
2.1 XAFS技术在单原子电催化水分解反应中的应用
电解水反应在储能和化工生产中具有广泛的应用.氢气生成反应(Hydrogen evolution reaction,HER)是电解水反应的阴极半反应,因具有现实的应用前景和简单的反应机理而成为最早得到广泛关注和充分研究的电化学反应之一.Pt基金属催化剂是HER反应最常用的催化剂,已得到大规模商业化应用,但Pt的高昂价格使得降低HER催化剂的成本成为现代工业生产中的一个重大课题.近年来,单原子催化剂因其均一的活性位点结构和极高的原子利用率成为了HER催化剂研发的焦点之一[33~35],而XAFS技术也在这些催化剂的结构表征和机理探究中发挥了关键的作用.
Fang 等[22]采用原子层沉积法(Atomic layer deposition,ALD)在以Uio-66-NH2MOF 材料为模板构建的氮掺杂多孔碳材料衬底上成功负载了均匀分布的Pt 单原子位点,构筑了一种新型HER 电催化剂(图6),在大幅降低Pt载量(Pt载量仅2.5%)的同时获得了超越商业Pt/C催化剂的HER活性(酸性反应条件下电流密度10 mA/cm2时过电位仅19 mV,相同条件下商用Pt/C 催化剂过电位为25 mV).借助EXAFS拟合技术,他们判明该催化剂中的Pt原子的近邻配位原子主要为C原子或N原子,配位数为4,且不存在Pt-Pt配位.他们还结合DFT理论计算和XANES白线峰面积分析,精确推断出催化剂中Pt的存在形式为Pt1-C3N1.随后,他们开展了电化学原位XAFS测试,观测了不同电压下XANES白线峰的面积变化情况,并对不同电压下获取的EXAFS曲线进行了拟合,发现在一定的阴极电压下,Pt与衬底连接的配位数显著降低[图6(D)],且化合价降低[图6(B)],形成一种更利于H原子吸附的“近自由”状态,从而显著提高了催化剂的性能.
近年来,除了Pt基催化剂外,一些非贵金属基的HER单原子催化剂也得到了长足的发展.非贵金属基催化剂的应用可以进一步降低催化剂的成本,有望带来更为深刻的产业革命.Cao 等[36]以磷化C3N4为衬底,用浸渍法成功负载了Co单原子位点,成功获得了具有相当可观的碱性HER活性(电流密度为10 mA/cm2时过电位为89 mV)的纳米催化剂.他们以此催化剂为基础开展了丰富的原位XAFS实验,精确表征了催化剂在HER反应中的活性位点结构变化(图7).借助EXAFS拟合,他们发现在未加电压时,催化剂的活性位点结构为Co1-N4,且包含2个较短的Co—N键(0.206 nm)和2个较长的Co—N键(0.218 nm).借助ΔXANES 曲线的解析[图7(A)~(C)],他们发现在施加了HER 阴极电压后,Co 的价态升高,又结合EXAFS拟合[图7(D)],发现Co—N/O配位结构的配位数发生变化.以此为基础,在DFT理论计算和近边模拟计算的帮助下,他们发现了在阴极电压下Co1-N4结构向Co1-N2O1或Co1-N2O2结构转化的过程,并证实了催化剂在阴极电压下形成的高价态HO-Co1-N2结构对H2O分子的强吸附作用是该催化剂优异的碱性HER活性的来源.
氧气生成反应(Oxygen evolution reaction,OER)是电解水制氢反应的阳极半反应,与HER反应相互对应.然而,与HER不同,大部分Pt基催化剂和其它贵金属基催化剂在OER反应端的活性一般[37~39],而基于化学反应动力学火山峰预测研制的各种金属氧化物催化剂又难以在实际工业生产条件下稳定工作,这使得OER 催化剂的研发与HER 催化剂相比相当滞后.过渡金属基单原子催化剂可以很好地兼顾催化剂的质量活性和稳定性,近年来在OER反应中得到广泛的关注.而XAFS技术因其对单原子催化剂强大的表征能力,在这种新式催化剂的作用机理阐明中发挥了关键的作用.
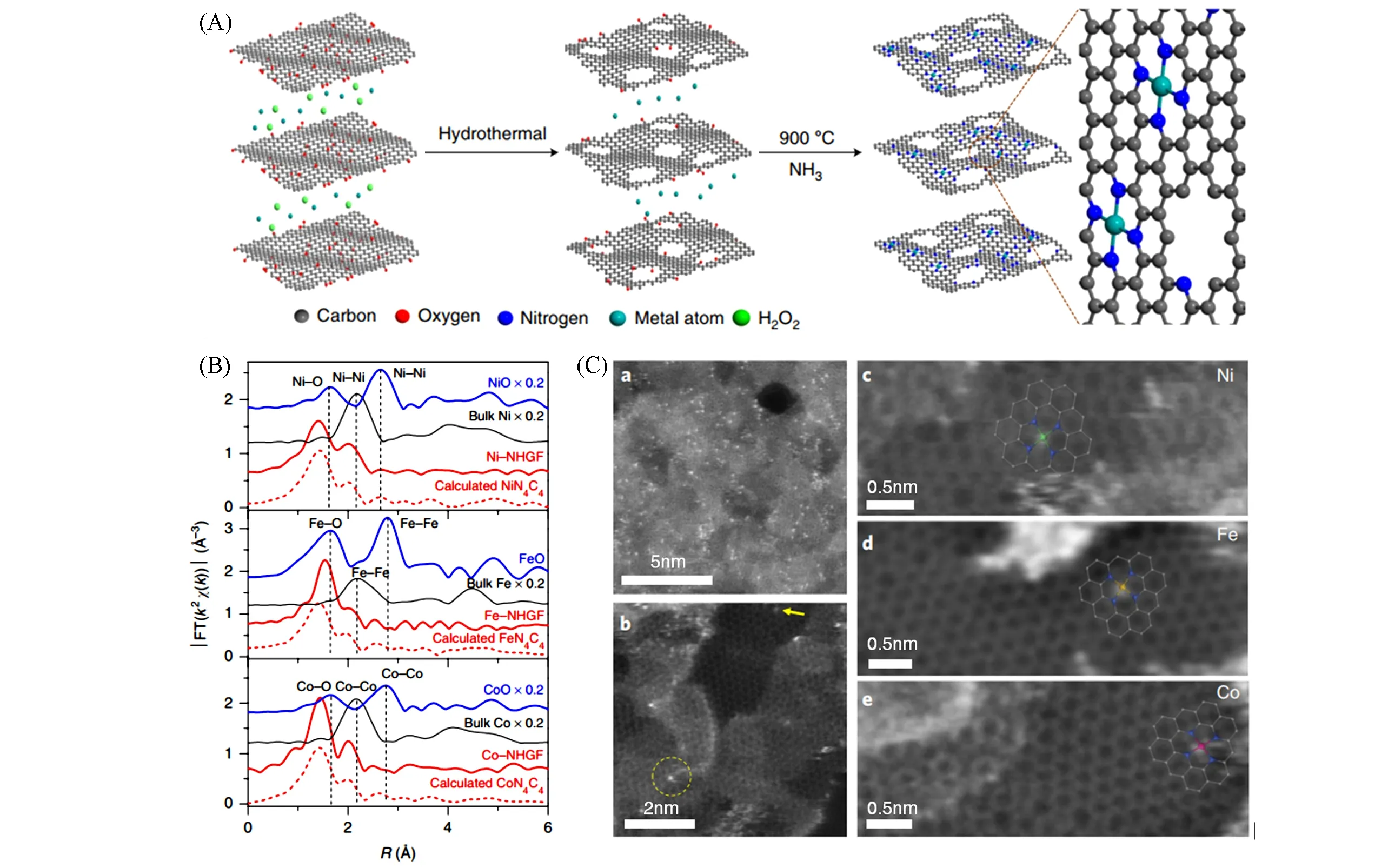
Fig.8 Illustration of the series M⁃NHGFs[40]
Fei 等[40]提出了一种普适的单原子催化剂合成方法[图8(A)],制备了一系列M-NHGF 催化剂(M=Fe,Co,Ni,NHGF=nitrogen-doped holey graphene framework).这些催化剂均具有可观的碱性OER活性(性能最佳的Ni-NHGF催化剂在10 mA/cm2电流密度下的过电位达331 mV,和商用RuO2/C催化剂接近)和稳定性.结合EXAFS 拟合[图8(B)]和XANES 模拟计算,他们精确表征了这些催化剂金属位点的近邻结构,发现它们都具备独特的M-N4C4O 结构,并在高倍率TEM 图像上找到了直观的证据[图8(C)].他们还结合DFT理论计算,探索了这一系列催化剂的作用机理,并对Ni-NHGF最优的性能给出了合理的解释.这一工作为普适性OER单原子催化剂的合成与机理描述提供了全新的思路.
Cao 等[41]在磷化C3N4(PCN)上构筑单原子Ru 位点,成功合成了一种新型OER 催化剂[图9(A)和(B)].该催化剂在0.5 mol/L H2SO4中表现出相当出色的OER 活性(电流密度达到10 mA/cm2时的过电位为267 mV,优于商用RuO2/C催化剂)和可观的工作稳定性(在1.5 Vvs.RHE工作条件下工作30 h性能仅衰减5%).借助EXAFS 拟合[图9(D)],他们确认了该催化剂中Ru 的存在形式为Ru1-N4,而没有Ru—O,Ru—Cl和Ru—Ru键存在.此外,他们还开展了原位XAFS和原位红外光谱(SR-IR)实验,经由不同电压下XANES吸收边位置变化的追踪和不同电压下EXAFS曲线的拟合,结合原位红外光谱的吸附物种特征峰分析,发现Ru1-N4结构在OER 工作电位下会首先吸附一个O 原子形成O-Ru-N4中间体,然后再参与常规的OER电化学循环[图9(F)],并通过DFT理论计算对这一机理进行了验证.

Fig.9 Illustration of single⁃atom Ru electrocatalyst for OER[41]
2.2 XAFS技术在单原子催化燃料电池反应中的应用
质子交换膜燃料电池(Proton exchange membrane fuel cells,PEMFCs)作为一种零碳排放的新型供能系统,近年来在新能源汽车领域得到广泛的应用.PEMFCs 的阴极反应为氧气还原反应(Oxygen reduction reaction,ORR),阳极反应为氢气氧化反应(Hydrogen oxidation reaction,HOR).目前绝大部分的实用PEMFCs 以Pt 基催化剂催化ORR 和HOR 反应.但是,Pt 的价格相当高,使催化剂成本占PEMFCs总成本的比例高达41%,其中ORR反应端缓慢的动力学使其在PEMFCs系统的搭建中消耗掉约80%的Pt[42],严重制约了PEMFCs的低成本商业化发展.因此,发展低Pt载量甚至非贵金属基的高活性ORR催化剂在近年来成为了产业界和学术界共同关注的热门课题,而单原子催化剂极高的原子利用率和独特的化学活性使其在ORR反应中具有广阔的应用前景.由于单原子Pt催化剂在ORR反应中会催化氧气的部分还原,生成对燃料电池有毒化作用的H2O2[43],对ORR 单原子催化剂的研制集中在Fe、Co 等过渡金属上[44~47].XAFS 技术在这些材料配位结构的判别和作用机理的阐释中发挥了重要的作用.
Mehmood等[48]结合分步浸渍和两步煅烧,成功在以ZIF-8为模板的氮掺杂多孔碳上负载了载量高达7%的Fe单原子位点[图10(A)],获取的催化剂表现出优异的ORR性能(酸性反应条件下半波电位高达0.815 V).他们还以此催化剂为基础组装单片PEMFC 进行测试,证实了该催化剂的商业应用前景.通过EXAFS[图10(B)和(C)]和穆斯堡尔谱[图10(D)和(E)]的综合分析,他们深入阐释了Fe物种在经历了高温活化过程后近邻结构由Fe-N、Fe-O复杂混合配位转化为Fe-N4均匀构型的变化过程,为ORR单原子催化剂的合理设计提供了全新的思路.
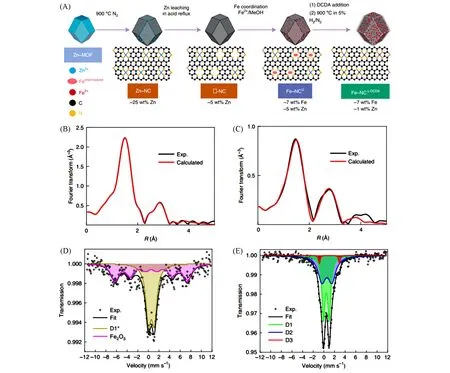
Fig.10 Illustration of the high⁃loaded single⁃atom Fe catalyst[48]
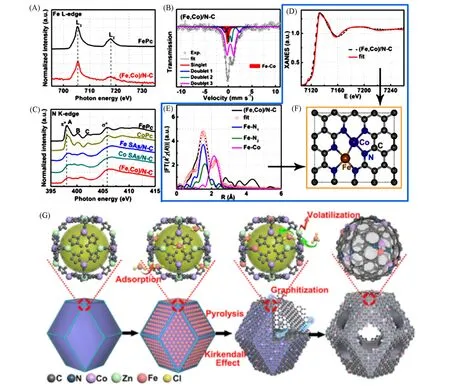
Fig.11 Illustration of(Fe,Co)/NC catalyst[49]
近年来,双金属位点的ORR单原子催化剂同样得到了广泛的关注.Wang等[49]创新性地发展了一种“主-客”合成策略,利用Zn1Co1-BMOF 中的Co 吸附浸渍液中的Fe,再通过高温条件除去Zn,制备了具有Fe-Co双金属位点的特殊单原子催化剂[图11(G)].该催化剂表现出优异的ORR活性(酸性条件下半波电位0.858 V)和工况稳定性(电流密度1000 mA/cm2条件下稳定工作100 h无明显失活).结合软线XAS测试[图11(A)和(C)]、穆斯堡尔谱[图11(B)]、EXAFS拟合[图11(E)]和近边计算[图11(D)],他们成功区分了Fe-N与Fe-O,Fe-Fe与Fe-Co,Co-N与Co-O等单纯EXAFS 拟合难以区分的配位结构,并以拟合结果为出发点构建了Fe-Co双原子位点的空间构型[图11(F)],此外,他们还结合DFT理论计算对该双原子位点的作用机理进行了深入阐释.
2.3 XAFS技术在单原子催化二氧化碳还原反应中的应用
电化学二氧化碳还原反应(Carbon dioxide Reduction Reaction,CO2RR)是将温室气体CO2在电化学条件下转化为CO,CH4,HCOOH 及C2H5OH等燃料和工业产品的反应,近年来因其诱人的新能源领域应用前景得到广泛的关注[50~52].然而,与研究较为充分的HER,OER 和ORR 等电化学反应相比,CO2RR催化剂的研制面临诸多困境,包括CO2分子的高稳定性和低水溶解性、CO2RR反应的机理复杂性和产物多样性、竞争性HER反应的影响等.单原子催化剂的催化位点结构单一,质量活性高,稳定性优异,非常适用于高选择性、高活性的CO2RR电化学反应,受到了广泛关注.在这些催化剂的结构表征中,XAFS技术同样发挥了不可或缺的作用.
Ding 等[20]以设计的Ni2(dppm)2Cl3团簇为金属前驱体,以ZIF-8 衍生的氮掺杂多孔碳为载体,实现了结构明确的碳基Ni2双原子位点催化剂的精确构筑(图12).他们发现该“双原子”催化剂在CO2电化学还原反应中表现出优异的活性和选择性(碱性条件下稳定工作电流密度150 mA/cm2,CO法拉第效率高达97.1%).他们将近边计算[图12(C)]与EXAFS拟合[图12(B)]结合,解决了单纯的EXAFS拟合无法区分N 与O 配位的问题,清晰地揭示了Ni2位点在电催化CO2还原反应中原子和电子结构的动态演变.研究结果表明,初始的Ni2-N6结构吸附电催化环境中的含氧官能团,从而形成O-Ni2-N6结构,同时在反应过程中伴随着Ni原子间距缩短和相互作用增强.DFT理论计算表明,经过动态演变形成的关键O-Ni2-N6结构会显著降低CO2分子活化的能垒,使该催化剂表现出优异的性能.
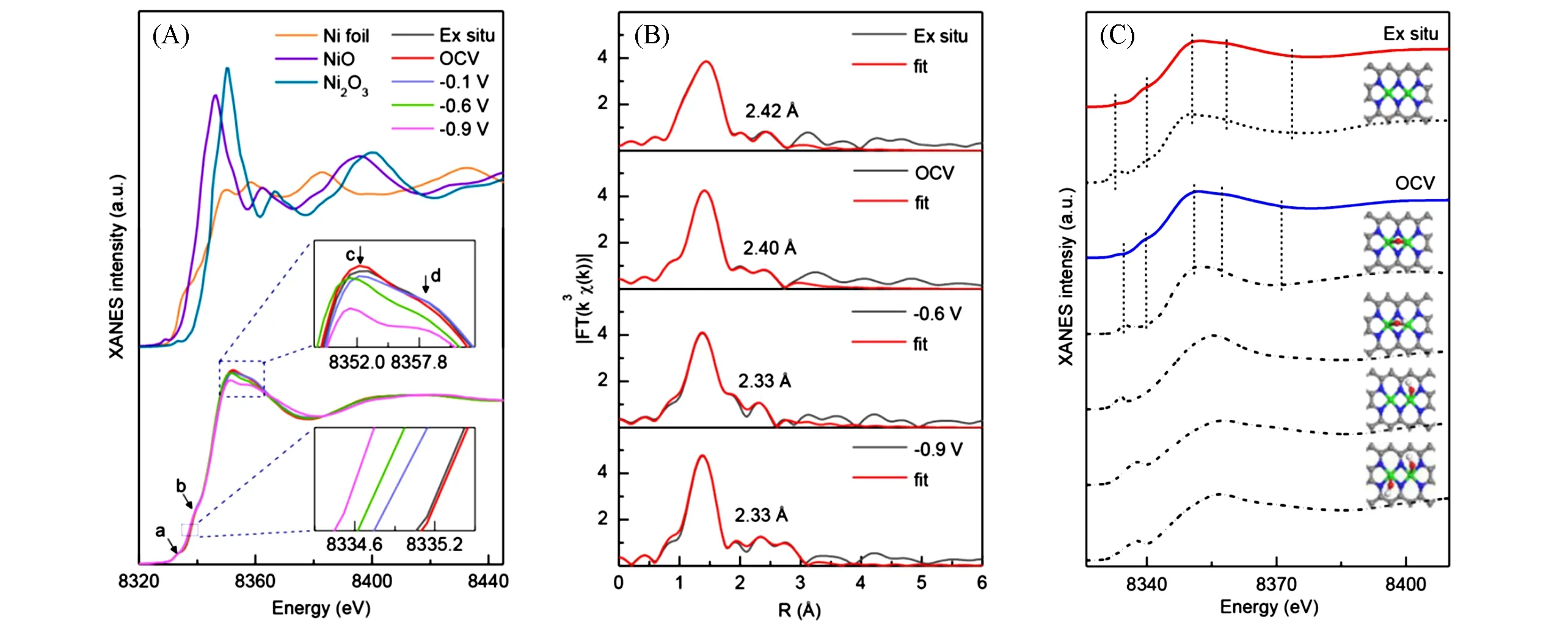
Fig.12 Illustration of Ni dinuclear site catalyst(Ni DSC)[20]
Liu 等[53]在油胺乙二醇体系中制备了Cu1/Au 单原子合金催化剂,并以此为模型催化剂,借助细致全面的原位XAFS分析[图13(A)~(C)],深入探究了Cu原子在CO2RR工况条件下配位结构的变化.他们对不同电压下获取的EXAFS曲线进行拟合,探测到Cu-Au配位数在还原电位下的变化情况,并以此为基础进行Material Studio 建模,发现Cu 原子在还原电位下由Au 多面体纳米颗粒的顶角位置迁移到(100)面内,并结合DFT理论计算阐明了这一变化对Au纳米颗粒电子结构的调节作用[图13(D)].此工作为单原子合金催化CO2RR反应机理的研究提供了全新的思路.
3 总结与展望
XAFS技术是一种20世纪80年代开始迅速发展起来的材料表征技术,具有对中心吸收原子的局域结构和化学环境敏感的特征,能够在原子尺度上给出某一原子周围几个邻近配位壳层的结构信息.经过半个多世纪的发展完善,研究者已开发出丰富多彩的XAFS原位实验方法,可以满足各种复杂化学条件的样品测试.XAFS技术的低浓度、低结晶度样品探测能力以及其独特的对待测元素短程有序结构信息的高度敏感性使其非常适合探测单原子催化剂的局域电子结构和空间结构,为待测样品活性中心位点的结构明确提供关键信息.此外,XAFS技术对样品的低破坏性使其适用于多种原位电化学测试,可以用于探测单原子催化剂在多种电化学工况条件下的结构变化,为催化剂作用机理的阐明提供间接证据.因此,近年来,XAFS 技术被广泛应用于HER、OER、ORR、CO2RR 等多种单原子催化剂参与的电化学反应中,取得了丰硕的研究成果.
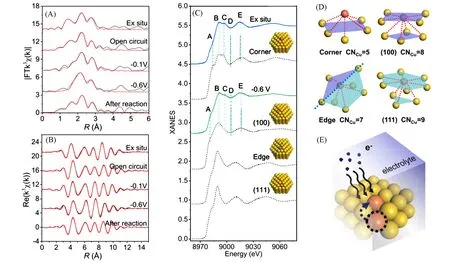
Fig.13 The in situ XAFS analysis of Cu1/Au catalyst[53]
目前,XAFS技术在单原子电催化领域已得到广泛的应用.但由于单原子催化剂的目标元素含量往往较低,且水相电解液对荧光信号的影响相当明显,大部分的电化学原位XAFS实验获取的数据信噪比较低,分析处理繁琐复杂,且往往具有歧义.针对这一问题的解决思路主要有:(1)进一步优化电化学原位XAFS测试的实验装置,尽可能降低电解液吸光作用和其余干扰因素的影响.目前已有相当多的研究者在进行XAFS测试时倾向于自行设计或定制专用的原位反应池[54],以满足自己课题的独特测试需求;(2)发展新一代光源,从本质上提高入射X 光的通量,从而提高XAFS 信号的信噪比.目前,瑞典、美国等国均有第四代光源设施,中国北京、合肥两地的第四代光源建设项目也已立项.有了这些全新的大型科学装置的支持,单原子催化领域中的XAFS测试与分析有望得到全面的优化.
此外,XAFS技术在单原子催化领域的应用还有广阔的拓展前景:(1)XAFS线站的自动化与智能化.目前大部分的XAFS线站手动操作部分多,调试方式简单原始,且难以进行大批量样品的测试.未来,在XAFS线站引入自动化换样、机器学习数据处理等新技术,一定能大幅提高XAFS测试效率和质量,使其在单原子催化领域得到更广泛的应用;(2)原位XAFS 和其它原位谱学表征的联用.原位XAFS技术与原位红外光谱、原位拉曼光谱、STXM成像等多种原位实验技术的联用能为单原子电催化剂的作用机理提供更全面的信息.然而,不同谱学表征手段的实验条件不同,如何在相同的电化学反应条件下进行多模式原位实验,仍是一大挑战;(3)时间分辨XAFS技术在单原子电催化领域的应用.现有的较成熟的时间分辨XAFS技术,无论是ED-XAFS还是Pump-probe XAFS,都非常不适合单原子催化剂的原位表征.这使得现有的单原子电催化研究很难涉及µs乃至ns时间尺度,单原子电催化剂的作用机理、表面中间体的吸附和解离等动态过程也难以得到明确.未来,开发新的时间分辨XAFS 技术或优化已有的ED-XAFS和Pump-probe XAFS,以实现更方便、更实用、与实际催化体系更贴合的ms以下时间尺度的XAFS探测,必将成为单原子电催化原位表征领域的一个重要课题.
猜你喜欢
机电安全(2022年5期)2022-12-13
实用手外科杂志(2022年2期)2022-08-31
陶瓷学报(2021年5期)2021-11-22
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
科学(2020年1期)2020-01-06
当代陕西(2019年6期)2019-04-17
天然产物研究与开发(2018年5期)2018-06-13
中国卫生(2015年12期)2015-11-10
中国卫生(2015年10期)2015-11-10
外语学刊(2014年3期)2014-12-03
