
单原子催化剂的合成及其能源电催化应用的研究进展
2022-09-19 06:28俞志勇黄小青
高等学校化学学报 2022年9期
姚 青,俞志勇,黄小青
(1.厦门大学化学化工学院,厦门 361005;2.苏州大学材料与化学化工学部,苏州 215123)
自工业革命以来,与日俱增的化石燃料消耗使地球资源与环境面临着巨大的压力,能源问题已成为人类未来发展面临的重大挑战之一.如今亟需开发绿色、清洁的可再生能源以减少对化石燃料的依赖[1,2].近年来,风能、太阳能、潮汐能等可再生能源的利用技术得到快速的发展,然而该类能源固有的间歇性、地域性和波动性等问题仍未得到真正的解决[3~5].因此,完善可再生能源的存储和转化技术是实现全球能源转型的关键.其中,将间歇性可再生能源转化为电能来驱动电化学反应,将环境中的二氧化碳、氮气和水等小分子转化可储存的燃料或有价值的化学品,被认为是一条有前景且可持续的路径[6~18].但实际的电化学反应往往面临着反应动力学缓慢、稳定性不佳、选择性低及成本高等现实挑战,在很大程度上限制了该技术在实际生产中的推广与应用[2,19~22].因此,开发高性能且低成本的电催化剂,对于推动可再生能源存储与转化技术的发展具有重要意义.
“单原子催化”的概念最早由张涛院士、李隽教授和刘景月教授[23]于2011年共同提出,他们报道了在FeOx上合成Pt单原子,并将单原子催化剂定义为所有金属组分均以单原子分散形式存在的负载型催化剂.相比于由成千上百个金属原子聚集而成的传统非均相催化剂,单原子催化剂达到了负载型催化剂的极限,极大地提高了在非均相催化中原子的利用率[24~28].另外,单原子催化剂在尺寸上的急剧减小,引起了许多不同于传统催化剂的特性(如不饱和的配位环境、强金属-载体相互作用以及量子尺寸效应等[29~33]).得益于在物理、化学性质上的独特优势,单原子催化剂不仅能够在电催化反应中展现出“以一当十”的潜力,而且为阐明反应机理提供了理想的模型,从而为催化剂的合理设计提供了有效的依据[34~39].
本文综合评述了单原子催化剂的合成及其能源电催化应用的进展,介绍了单原子催化剂的常见表征手段,总结了单原子催化剂的合成方法(湿化学法、高温热解法、原子沉积法及电化学沉积法等),并介绍了该类材料在氧还原、二氧化碳电还原、电解水和氮气电还原反应中的研究进展,重点探讨了催化剂微观结构与其性能之间的关系,最后对单原子能源电催化领域所面临的挑战进行了总结,并对该领域未来的发展方向进行了展望.
1 表征手段
鉴定原子级分散的金属并确定其分布、配位环境及与载体之间的相互作用,对于研究单原子催化剂至关重要[40~44].随着原子分辨技术与同步辐射技术的快速发展,单原子催化剂在不同催化领域的催化机理以及构效关系有望得到更深入的了解.如,球差校正扫描透射电子显微镜(Aberration Corrected Scanning Transmission Electron Microscopy,AC-STEM)能够直接观察到锚定在载体上的单个金属原子的图像,是目前单原子催化剂研究中不可或缺的表征手段[45].Yuan 等[46]利用AC-STEM 清晰地观察到亮度更高的Fe 单原子均匀分布于碳载体上[图1(A)],同时借助电子能量损失谱(Electron Energy Loss Spectroscopy,EELS)以及能量色散X 射线元素映射(Energy Dispersive X-ray Spectroscopy,EDX)等表征手段,得到了原子水平上的元素组成和分布情况[图1(B)和(C)].除了碳载体,AC-STEM技术还可用于检测金属合金载体或其它含金属载体的单原子[47~49].然而,电子显微技术的可视范围有限,只能选择特定的局部区域.同步辐射X射线吸收光谱(X-ray Absorption Spectroscopy,XAS)则是一种更强大的技术,可以通过阐明单原子的电子和几何结构,了解载体上单原子的结构信息[50].这一技术根据作用机理与峰形可分为X射线吸收近边结构(X-ray Absorption Near Edge Structure,XANES)与X射线吸收精细结构谱(X-ray Absorption Fine Structure,EXAFS)[51].对于EXAFS,通常能够从其谱图中得到相邻原子的种类、配位数以及键长等信息,而XANES技术则对价态与电荷转移信息更为敏感,因此常用于区分化学元素价态.Li 等[52]通过研究EXAFS 谱图发现,Ir 单原子以Ir-N4结构分布于氮掺杂碳中[图1(D)],同时从其XANES 谱图可知,Ir单原子的平均价态介于金属Ir与IrO2之间[图1(E)].此外,还可以通过原位XAS技术研究催化过程中金属价态的变化情况.如[图1(F)]所示,随着作用电位的增加,可观测到Ir单原子的平均价态从4.81减小至1.68.除了上述的AC-STEM 和XAS 这些可以直观地给出单原子位点信息的技术外,原子探针层析(Atom Probe Tomography,APT)技术、红外光谱(Infrared Spectroscopy,IR)技术、氢气程序升温还原(H2Temperature-Programmed Reduction,H2-TPR)等技术,在单原子催化剂的结构分析中也起到重要的辅助作用.如APT 能够实现对材料的原子级三维(3D)成像及组分探测,尽管APT不能提供单个原子的确切结构,但该技术可以有效地辅助STEM与XAS以提供更真实的原子结构信息[21].如,Jiang等[53]成功利用APT技术表征了锚定在碳纳米管(Carbon Nanotube,CNT)上的单个Fe原子.从重建层析图像在二维(2D)空间的投影图中可以清晰地观察到,以绿色突出显示的Fe单原子均匀地分布在样品中[图1(G)].从碳原子分布的二维等高线图可以得到碳纳米管载体的形状[图1(H)].对较小的局部区域进行分析可知,Fe原子沿着碳纳米管均匀分布,并且没有观察到距离小于3 Å(1 Å=0.1 nm)的相邻Fe原子,证实了Fe原子在碳纳米管中的原子级分散[图1(I)].几个原子层的侧视图及俯视图揭示了Fe单原子可能与相邻的C和O形成Fe-C-O的配位形式[图1(J)].原位红外光谱(in situIR)可以分辨吸附在催化剂表面探针分子的振动模式,因而可用于区分单原子位点和纳米颗粒位点的结构差异[38].如,Wang 等[54]利用原位漫反射红外傅里叶变换光谱(in situDiffuse Reflectance Infrared Fourier Transform Spectroscopy,in situDRIFT)研究了CO 在PdNi 合金单原子催化剂表面的吸附方式.位于2092和1975 cm−1处的峰分别对应CO的线式吸附与桥式吸附.在5%Pd-Ni/SiO2中,2092 cm−1处的峰明显强于1975 cm−1处的峰,证明了其中的Pd 主要以单原子形式存在[图1(K)].另外,随着材料中Pd的相对含量增加,1975 cm−1处的CO吸附峰明显增强,说明在Ni金属载体中连续的Pd 原子比例增多.H2-TPR 技术可以提供单原子催化剂在还原过程中氢气消耗量与还原温度等信息,因而可以根据测试谱图获得一些结构参数.如,通过谱图中还原峰的偏移可以推断单原子的价态、周围环境以及与载体的相互作用强度等[55,56].
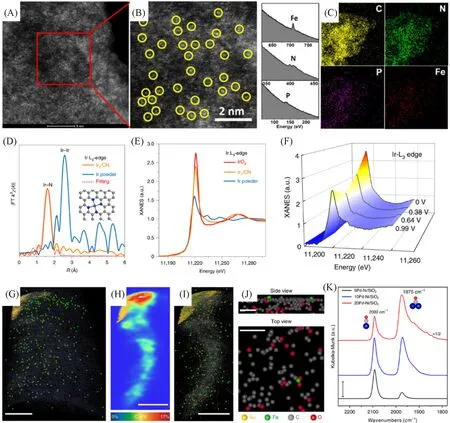
Fig.1 AC⁃STEM(A)and enlarged images of Fe⁃N/P⁃C⁃700 and EELS atomic spectra from the bright dots shown by the yellow circles(B),EDS elemental mappings of Fe⁃N/P⁃C⁃700(C)[46],FT⁃EXAFS spectra of Ir1/CN and iridium powder(D),XANES of Ir1/CN,iridium powder,and IrO2(E),iridium L3⁃edge XANES spectra of Ir1/CN at various potentials during the potentiostatic FAOR(F)[52],reconstructed APT data for a region containing Fe⁃CN(G) with a 2D contour plot of C atomic concentration(H)and the corresponding reconstruction for a region of 40 nm×40 nm×80 nm surrounding a CNT(I),side and top view of CNT planes(J)[53],DRIFTS CO chemisorption of the xPd⁃Ni/SiO2 samples(x=5,10,and 20)at the CO saturation coverage(K)[54]
2 合成方法
制备具有高负载量和具有明确局部配位环境的单原子催化剂对于工业化应用具有十分重要的意义.然而,与传统的催化剂相比,单原子催化剂的合成是一个具有挑战性的问题,特别是大规模、高负载量的单原子催化剂的高效制备尤为困难[57~60].随着单原子催化领域的不断发展,研究者们已经开发了多种合成方法(湿化学法、高温热解法、原子层沉积法、电化学沉积法等)来制备单原子催化剂.
2.1 湿化学法
湿化学法包括共沉淀法、浸渍法及离子交换等.由于操作简单、容易实施等优点,被认为是目前实现单原子催化剂合成最基本和最有效的方法[34,61~63].在湿化学法制备单原子催化剂的过程中,首先需要将金属前驱体锚定在载体上,然后选择合适的处理方式将不需要的配体去除,再进行煅烧或还原等过程.如,Qiao 等[23]首次采用了共沉淀的方法,制备出锚定在FeOx上的Pt 单原子(Pt/FeOx);Lin 等[64]利用类似的方法得到了锚定在FeOx上的Ir 单原子催化剂(Ir/FeOx).他们首先将贵金属前驱体的水溶液与硝酸铁溶液在碱性条件下进行混合,得到的沉淀物经过过滤、洗涤和煅烧后,可得到Pt/FeOx和Ir/FeOx.由于较高的表面能和低配位环境,金属单原子倾向于团聚成团簇或者纳米颗粒,因此,在湿化学法中合成的单原子载量通常较低,且需要表面积较大的载体来锚定单原子.值得注意的是,金属与载体之间的强相互作用,是防止单原子发生团聚的一大关键因素.因此,载体上的表面性质至关重要.Wang等[65]以具有介孔结构的S掺杂C(meso_S-C)为载体,通过浸渍法制备了一系列贵金属单原子催化剂(M/meso_S-C;M=Pt,Pd,Rh,Ir).以Pt/meso_S-C 的合成为例,首先将meso_S-C 分散在去离子水中,再加入一定量的H2PtCl6水溶液;超声混合并剧烈搅拌后,进行旋蒸除水.最后将粉末在250 ℃下,5%H2-Ar的气氛中处理2 h(图2).研究发现,高S含量和高表面积的载体可以提供丰富的锚定位点并有效地防止金属原子聚集.此外,由于金属与S之间的强相互作用,在200~500 ℃的还原过程中,金属原子仍能稳定地存在.

Fig.2 Schematic illustration of the preparation and model structure of the atomically dispersed noble metal catalysts[65]
最近,Hai等[66]利用湿化学法在合成高载量单原子催化剂中取得了重大突破.他们开发了两步退火策略,用于大规模合成负载量高达23%的超高密度单金属和多金属的单原子催化剂(UHD-SAC).为了配体的可控去除,第一步的退火温度要低于金属前驱体的分解温度.随后,第二步在较高温度下进行退火以去除多余的配体,从而将化学吸附的金属前驱体转化为具有明确结构的UHD-SAC[图3(A)].机理研究表明,通过逐步去除配体来控制金属前驱体与载体的键合,可以有效地防止金属前驱体在热处理过程中发生团聚.此外,该方法具有很好的普适性,可适用于V,Cr,Mn,Fe 等超过15种过渡金属和CeO2、氮掺杂的碳(NC,PNC)等这些具有不同特性的载体[图3(B)].更重要的是,该方法被成功地应用于自动化模式制备,以实现公斤级的制备,并呈现出很好的重复性,证明了该方法具有很好的可控性与高效性[图3(C)~(E)].

Fig.3 Strategy for the preparation of UHD⁃SACs(A),metal loadings achieved in this study on NC,PCN and CeO2 supports(B),photograph of the robotic synthesis platform and assignment of tools to unit operations(C),flowsheet of the synthesis protocol(D),comparison of metal content achieved by auto⁃mated and manual synthesis of Ni1/NC catalysts(E)[66]
2.2 高温热解法
高温热解法是合成单原子催化剂的常用策略之一[67].在合适的温度和气氛中对相应的前驱体进行热分解,可制备分散的单原子催化剂.由于金属有机框架(MOF)具有原子分散的金属节点和明确的有机配体,且具有高比表面积和丰富的孔结构,被认为是高温热解法中理想的模板或前驱体之一[68~71].近年来,由MOF 衍生的单原子催化剂的研究取得了很大进展.如,Yin 等[42]首次利用原位高温热解Zn/Co双金属MOF的方法,成功构建了稳定在氮掺杂的多孔碳上的Co单原子.在Zn/Co MOF中,Zn2+取代了部分Co2+,使得两个Co原子之间的距离适当扩大,由于Zn原子的沸点更低,在800 ℃以上的高温下会挥发,而高温热解生成的碳会还原位于金属节点的Co2+,从而得到Co单原子催化剂[图4(A)].但若目标金属原子不能作为MOF中的金属节点,则不能通过上述的方法制备相应的单原子催化剂.为了克服这一困难,研究人员发展了离子交换-热解策略.Zhao等[72]首次利用该策略制备了Ni单原子催化剂.他们首先将尺寸均匀的ZIF-8 分散于正己烷中,然后将Ni(NO3)2水溶液注入;经过混合后,Ni(NO3)2被限制在ZIF-8的孔道中;随后将该混合物在氩气中,1000 ℃的高温下进行热解.随着热解的进行,有机配体转变成了N掺杂的碳骨架.当热解温度高于900 ℃时,节点上的Zn发生蒸发,并随之产生周围富含N的缺陷位点,这些缺陷位点很容易被Ni2+占据.最终,孤立的Ni2+位点被N配位稳定并被碳还原,从而形成Ni单原子催化剂[图4(B)],类似的机制也被应用于合成Ru[43],Fe[73]等单原子催化剂.除了利用MOF的孔道限域效应,还可以通过目标金属取代部分MOF中金属节点的方法来构建单原子催化剂.如,Shan 等[74]以ZIF-67 为载体,以Na3IrCl6·xH2O 为Ir 的前驱体,实现了目标Ir 原子在ZIF-67中的取代型掺杂,经过热解后,最终得到了基于Co3O4晶尖石结构的Ir单原子催化剂[图4(C)].在高温热解法中,除了以MOF为模板/前驱体外,聚合物[75]、碳载体[76]和过渡金属化合物[48]等也被成功地应用于构建高质量的单原子催化剂.
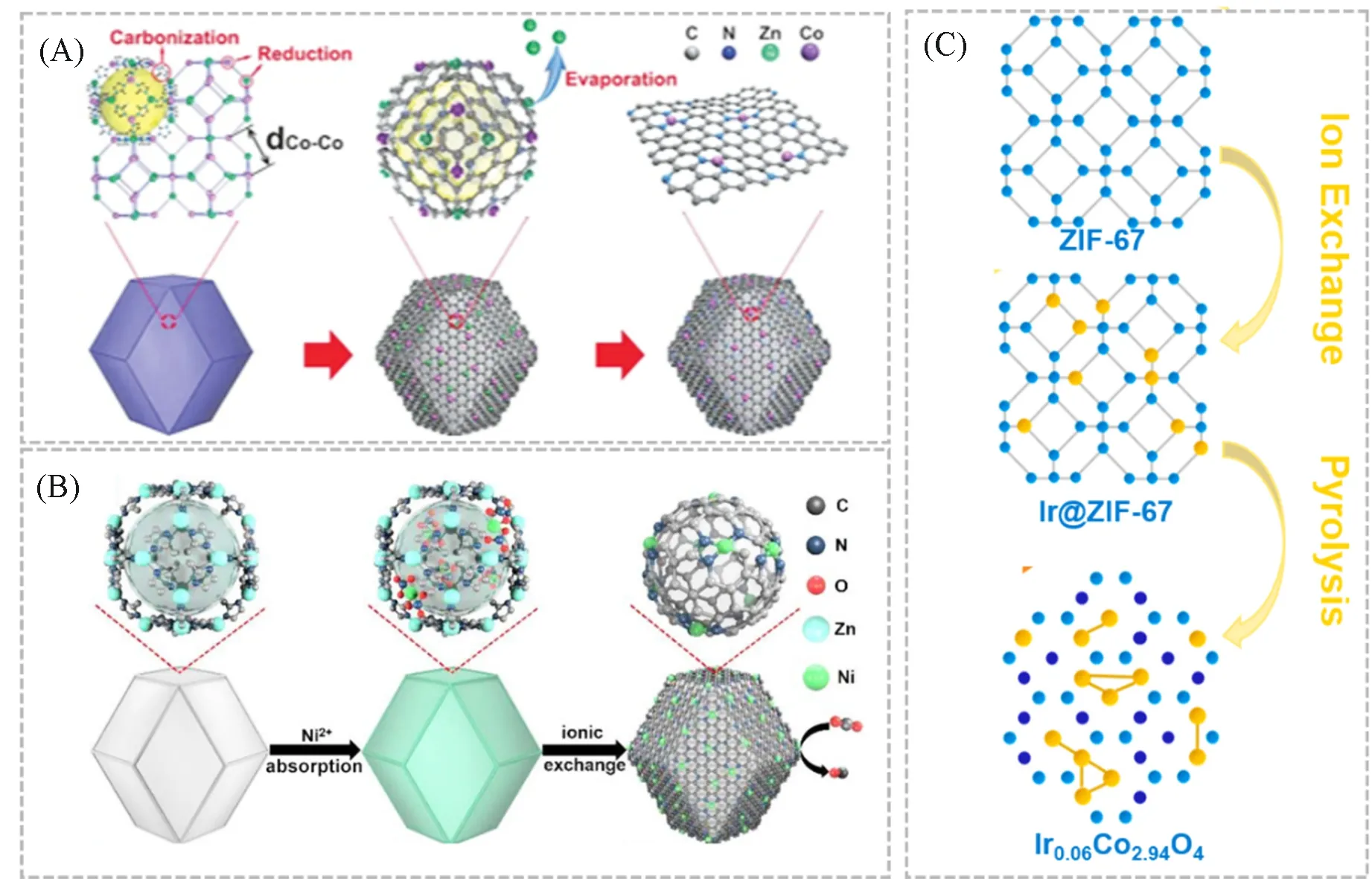
Fig.4 Formation of Co SAs/N⁃C(A) [42],scheme of the formation of Ni SAs/N⁃C(B) [72],formation of Ir0.06Co2.94O4(C)[74]
2.3 原子层沉积法
原子层沉积法(Atomic Layer Deposition,ALD)也被称为原子层外延法,是指将物质通过自限制的方式,以原子膜形式一层一层地精准沉积在载体表面上的催化剂合成技术[77~80].在原子层沉积法中,沉积参数可精确调控且重复性较好,能为负载型单原子催化剂的基础研究提供理想的模型.Li等[79]通过原子层沉积法,在石墨氮化碳衍生的氮掺杂碳纳米片(NCNS)上实现了高负载量的Pt 单原子催化剂.他们以三甲基(甲基环戊二烯基)-铂(MeCpPtMe3)和O2为前驱体,高纯度的氮气用作吹扫气和载气.制备过程中精确调控沉积的温度与MeCpPtMe3容器的温度,在NCNS 上进行一个循环的Pt ALD,可获得负载于NCNS上的Pt单原子(Pt1/NCNS)[图5(A)和(B)].研究发现,若同样以NCNS为载体,而以二茂钴[Co(Cp)2]为前驱体时,却只能得到Co的纳米颗粒或者团簇,这可能源于Co前驱体与载体间的相互作用过弱.基于此,Li 等[81]提出了将上述制备的Pt 单原子与ALD 相结合的方法,成功制备了Fe,Co,Ni 单原子催化剂.研究发现,在NCNS 上,Pt 单原子可以催化Co(Cp)2的解离,并使其以化学吸附的形式沉积在NCNS表面上,很好地促进了在ALD过程中形成稳定且分散的Co单原子[图5(C)].当采用相同的策略将前驱体换为Fe(Cp)2和Ni(Cp)2时,也成功得到了Fe,Ni单原子.不同于上述研究中以电负性较大的氮原子来锚定金属原子,Chen等[82]利用材料的本征缺陷作为锚定位点,以电负性较小的黑磷(BP)为载体,通过ALD制备了呈零价态的Pd单原子催化剂(Pd1/BP).在ALD中,他们首先在高温和真空氛围中去除黑磷表面的杂质并使表面的缺陷数量增加,然后将Pd的前驱体沉积到黑磷载体上与缺陷位点结合,最后通过臭氧处理将前驱体的多余配体去除,从而得到Pd 单原子催化剂[图5(D)].利用原子层沉积法制备单原子催化剂也存在一些明显的劣势,如产量较低,且成本高昂,在很大程度上限制了该方法的商业化应用.

Fig.5 Illustration for the synthesis(A) and representative aberration⁃corrected HAADF⁃STEM image of Pt1/NCNS(B)[79],illustration of Co ALD process on Pt1/NCNS(C)[81],schematic illustration of the synthesis of BP confined SACs via ALD(D)[82]
2.4 电化学沉积法
三电极体系的电化学沉积法操作简单,可在温和的条件下进行,也被应用于金属单原子催化剂的制备[83,84].在该方法中,金属原子的沉积速率至关重要,沉积速率过快而沉积位点不足时容易导致金属原子的聚集.另外,沉积的产物会受到电位、电流密度和酸碱度等参数的影响.Zhang等[85]首次通过电化学沉积法制备了Pt单原子催化剂.随后,Zhang等[86]发展了利用电化学沉积法制备单原子的普适性方法.仅通过改变金属前驱体和载体的种类,就可同时在阴极和阳极上沉积出30 多种单原子催化剂.以在Co(OH)2纳米片上沉积Ir单原子为例,Ir阳离子(IrCl3+)在电场的驱动下沉积在阴极的载体上[C-Ir1/Co(OH)2],Ir阴离子则沉积在阳极的载体上[A-Ir1/Co(OH)2][图6(A)和(B)].研究发现,前驱体浓度低于某一限度时,产物为单原子,而当前驱体浓度过高时,产物则有团簇或颗粒生成.有趣的是,由于阴极和阳极不同的沉积物种,以及阴极、阳极不同的氧化还原反应,制备得到的两极上的Ir单原子具有不同的价态和配位环境.从XANES谱图可知,C-Ir1/Co(OH)2中Ir单原子的价态降低,而A-Ir1/Co(OH)2中Ir单原子的价态升高[图6(C)].EXAFS谱图中Ir—Ir键的缺失证明了Ir以单原子的形式存在,不同的峰强度证明了在配位数上的差别[图6(D)].不同于上述常规的电化学沉积法,Shi等[87]提出了在二维硫化物的特异性位点上欠电位沉积金属单原子的新方法.以硫化物上分散的硫族原子为沉积位点,成功制备出了Pt单原子催化剂.该方法的优势之一在于欠电势沉积具有“自限制”特点,从而使单原子的形成不受沉积时间与前驱体浓度的限制[图6(E)和(F)].利用相同的方法,研究者们成功制备了Pd,Bi,Sn等单原子催化剂.

Fig.6 Schematic of cathodic(A) and anodic(B) deposition of Ir species,normalized XANES(C) and EXAFS(D) spectra at the Ir L3⁃edge for cathode⁃Ir1/Co(OH)2 and anode⁃Ir1/Co(OH)2[86],the mechanism of site⁃specific UPD(E,F)[87]
2.5 其它合成方法
除了上述方法外,质量分离-软着陆法、光化学法和固相熔融法等[26,88,89]也被应用于单原子催化剂的制备.其中质量分离-软着陆法是较为先进的物理沉积方法,该方法利用高频激光使金属气化,通过质谱仪精确调控不同尺寸的金属物种在载体表面沉积.然而,该方法条件苛刻、成本高昂且产量较低,难以实现单原子催化剂的规模化生产.因此,质量分离-软着陆法通常用于为基础研究提供理想的单原子模型催化剂.光化学法是指利用光能量来激活反应物而发生反应的一种方法.首先利用载体吸附金属前驱体,然后将相应的混合物置于光源下照射一段时间,以获得单原子催化剂.该方法具有制备过程简单、条件温和等优点,但制备得到的单原子催化剂通常负载量较低.在固相熔融法中,将金属前驱体与载体在合适的条件下研磨混合均匀后,置于高温下进行熔融,并结合相应的后处理方式得到单原子催化剂.固相熔融法的成本较低、工艺简单,但也存在固相之间反应性较差,对载体的热力学稳定性要求高等局限性.
3 能源电催化领域的应用
随着能源与环境问题的日益加剧,发展绿色能源存储与转化技术迫在眉睫.其中,与能源相关的电催化反应是近年来的研究热点[8,9,90].单原子催化剂由于其独特的物理、化学性质,在众多电催化反应都展现出很好的应用前景.相比于传统的非均相催化剂,单原子催化剂在理论上具有100%的原子利用率,这对于降低催化剂的成本具有非常重要的意义[26].另外,单原子催化剂的不饱和配位环境、与载体之间的强相互作用等特性,导致单原子中心电子结构的改变,这在很大程度上优化了电催化性能[91,92].下面选取了一些比较典型的电催化反应,介绍了单原子催化剂在能源电催化领域的应用和进展.
3.1 氧还原反应
燃料电池是一种能够将燃料和氧化剂中的化学能直接转换为电能的能量转化装置.其中四电子的氧还原反应(Oxygen Reduction Reaction,ORR)作为燃料电池的阴极反应,是目前热门的研究方向.Pt 基催化剂被认为是目前催化性能最好的ORR 催化剂[38,93~99].然而,Pt 储量稀少且价格昂贵,在一定程度上限制了它们的规模化应用.因此,在保证催化性能的同时,发展非贵金属或低载量贵金属ORR催化剂是研究者们亟需解决的难点.为了提高Pt在燃料电池中的利用率,Liu等[100]以富含碳缺陷的碳(BPdefect)为载体,制备出了超低Pt 载量(质量分数1.1%)的Pt 单原子催化剂(Pt1.1/BPdefect)并用于酸性ORR[图7(A)].测试结果表明,Pt1.1/BPdefect在0.1 mol/L HClO4中的表观ORR催化性能,优于负载在BP上但单原子占比较低的Pt催化剂(Pt1.1/BP),且与商业Pt/C的性能相当[图7(B)].理论计算结果表明,在碳双空位中由4个碳原子锚定的Pt单原子是主要的活性中心,该活性中心具有优异的四电子ORR的催化能力[图7(C)].此外,单原子Ru 催化剂也被用作高效的四电子ORR 催化剂[43,101].如,Xiao 等[43]通过高温热解法,合成了锚定在碳氮载体上的Ru单原子催化剂(Ru-SSC)[图7(D)],Ru,N,C呈现均匀分布的状态[图7(E)].EXAFS揭示了Ru单原子与4个氮原子配位,形成了Ru-N4结构[图7(F)].与Fe-SSC,Ru/N-C和Pt/C等材料相比,Ru-SSC具有最高的ORR性能,在0.8 V时,Ru-SSC的面积活性与质量活性分别达到了11.95 mA/cm2和4.78[图7(G)和(H)].值得一提的是,Ru-SSC具有优异的周期转化频率,达到4.99 e−·s−1·sites−1,明显高于多数已报道的催化剂[图7(I)].Pd 基催化剂被认为是最有希望替代Pt的ORR催化剂.然而,不同于传统的Pd基纳米催化剂,Pd单原子催化剂则对二电子ORR表现出较高的活性与选择性[102].
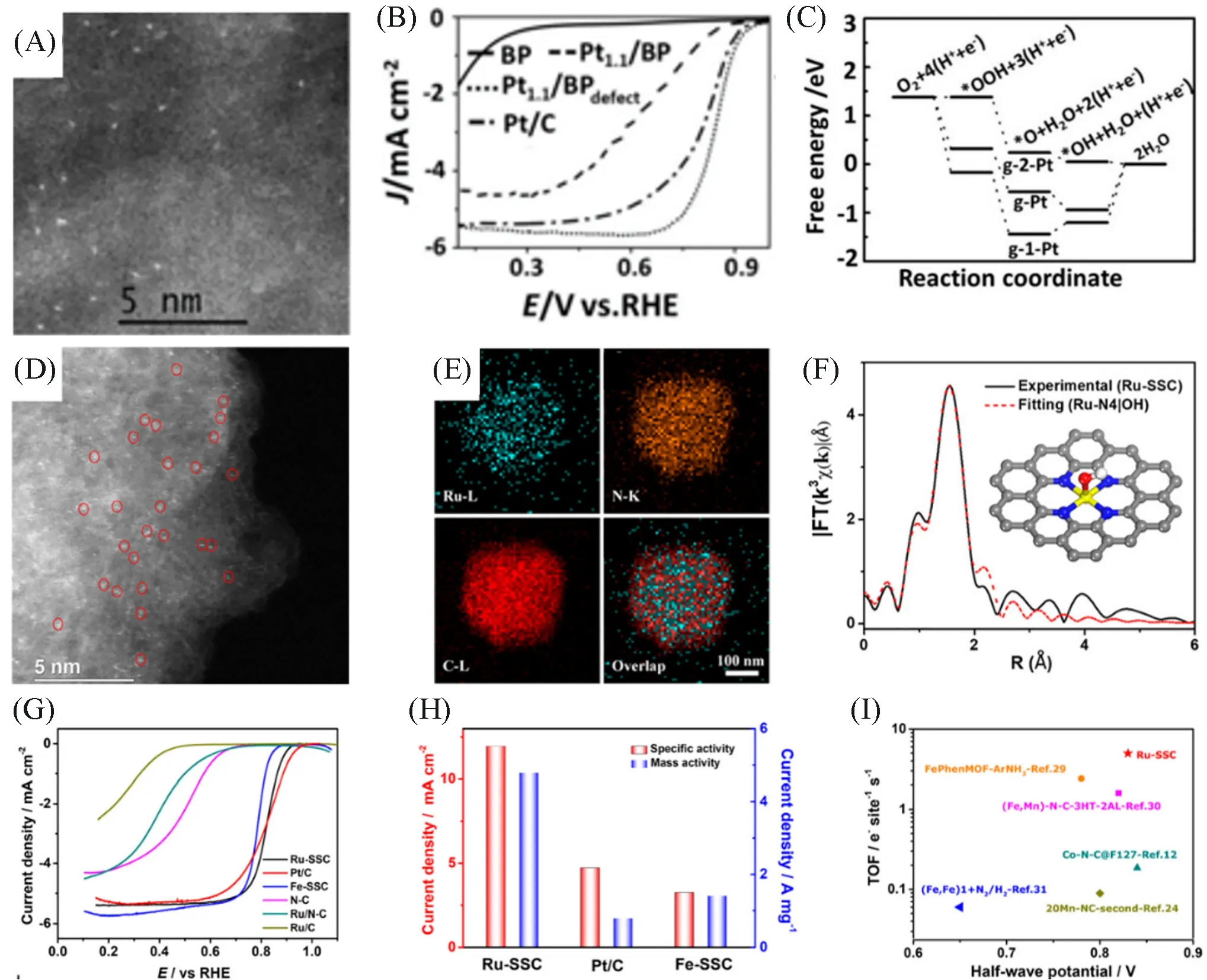
Fig.7 HAADF⁃STEM image of Pt1.1/BPdefect showing the dense distribution of Pt atoms on carbon(A),RDE polarization curves of BP,Pt1.1/BP,Pt1.1/BPdefect and the commercial Pt/C(B),free energy diagram for complete O2 reduction on single Pt atom supported on pristine graphene(g⁃Pt),single Pt atom suppor⁃ted on monovacancy graphene(g⁃1⁃Pt),and single Pt atom supported on divacancy graphene(g⁃2⁃Pt)substrates in acidic media at 0.83 V,respectively(C) [100],STEM image(D) and the corresponding elemental mappings(E)for the Ru⁃SSC,EXAFS fitting curve for Ru SSC(F),ORR polarization curves(G),specific activity and mass activity comparison among Ru⁃SSC,Pt/C,and Fe⁃SSC(H),TOF(mea⁃sured at 0.8 V vs. RHE)and E1/2 values of Ru⁃SSC and other recently reported SSCs(I)[43]
除贵金属单原子催化剂外,一些非贵金属单原子催化剂(如Fe[103~105],Co[106],Ce[107,108],Se[109],Sb[110]等)也被报道应用于催化ORR.Jin等[103]以水凝胶锚定策略在氮掺杂的碳基体上制备了密度可控的Fe 单原子催化剂(Fe-N4)[图8(A)],并揭示了位点之间的距离(dsite)与ORR 活性存在着很强的关联性.EXAFS 的拟合结果证明了Fe 单原子的存在,并揭示了Fe 与周围的4 个N 进行配位[图8(B)].随后,研究者们记录了不同Fe单原子的ORR极化曲线.由结果可知,电流密度随Fe单原子负载量的增加而增加[图8(C)].更有意思的是,当Fe单原子的负载量较低,即dsite大于1.2 nm时,催化剂的活性仅存在细微的差异.而当dsite小于1.2 nm时,相邻Fe-N4单元之间的强相互作用改变了电子结构,从而提高了ORR活性.而当相邻Fe单原子的dsite低于0.7 nm时,催化活性略有下降[图8(D)].
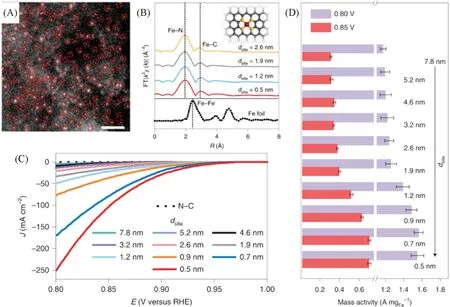
Fig.8 HAADF⁃STEM image of Fe⁃N4 SACs with dsite values of 0.5 nm(A),EXAFS spectra for four typical Fe⁃N4 catalysts with different dsite(B),LSV of the PPy⁃derived N—C and Fe SACs with different dsite values(C),MA normalized by the total weight of Fe in different samples according to the ICP⁃MS result(D)[103]
3.2 二氧化碳电还原反应
二氧化碳电还原反应(CO2Reduction Reaction,CO2RR)为生产化学燃料和化学品提供了一条有价值的途径,是缓解温室效应和实现碳中和目标的有效措施之一.然而CO2RR过程十分复杂,存在产物种类多、选择性低等问题[111~114].因此,开发高效的CO2RR催化剂以提升其活性与选择性是近年来的研究热点.单原子催化剂由于其独特的几何结构和电子性质,在CO2RR中也展示出了优异的催化性能.如,Wang等[115]通过简单的热处理方法,合成了锚定在C3N4上的Mg单原子催化剂(Mg-C3N4)[图9(A)和(B)],用于CO2还原生成CO.由于Mg原子具有离域化的s轨道,有效地克服了3d过渡金属位点CO脱附困难的瓶颈.相比于Fe,Co单原子催化剂(Fe-C3N4,Co-C3N4),Mg-C3N4展现出更高的CO选择性.通过程序升温脱附(TPD)可知,Fe-C3N4,Co-C3N4与CO 之间有很强的吸附作用,而Mg-C3N4上的CO 可以轻松地脱附[图9(C)].在H 型电解池的CO2RR 中,CO 的法拉第效率(FECO)可达到90%,明显优于Fe-C3N4,Co-C3N4等其它催化剂[图9(D)].Mg-C3N4的Tafel斜率为99.6 mV/dec,表明其决速步为*COOH的形成,而Fe-C3N4,Co-C3N4的Tafel 斜率分别为344.1 和258.2 mV/dec,表明两者的决速步骤为CO 的脱附[图9(E)].在流动型电解池的测试中,保证FECO≥90%的同时,电流密度可达到300 mA/cm2[图9(F)].除此之外,Mn[116],Cd[117],Ni[118,119],Pd[120]等单原子催化剂也被报道用于高效的CO2RR生成CO,而Zn单原子催化剂则对CH4具有较高的选择性[121].
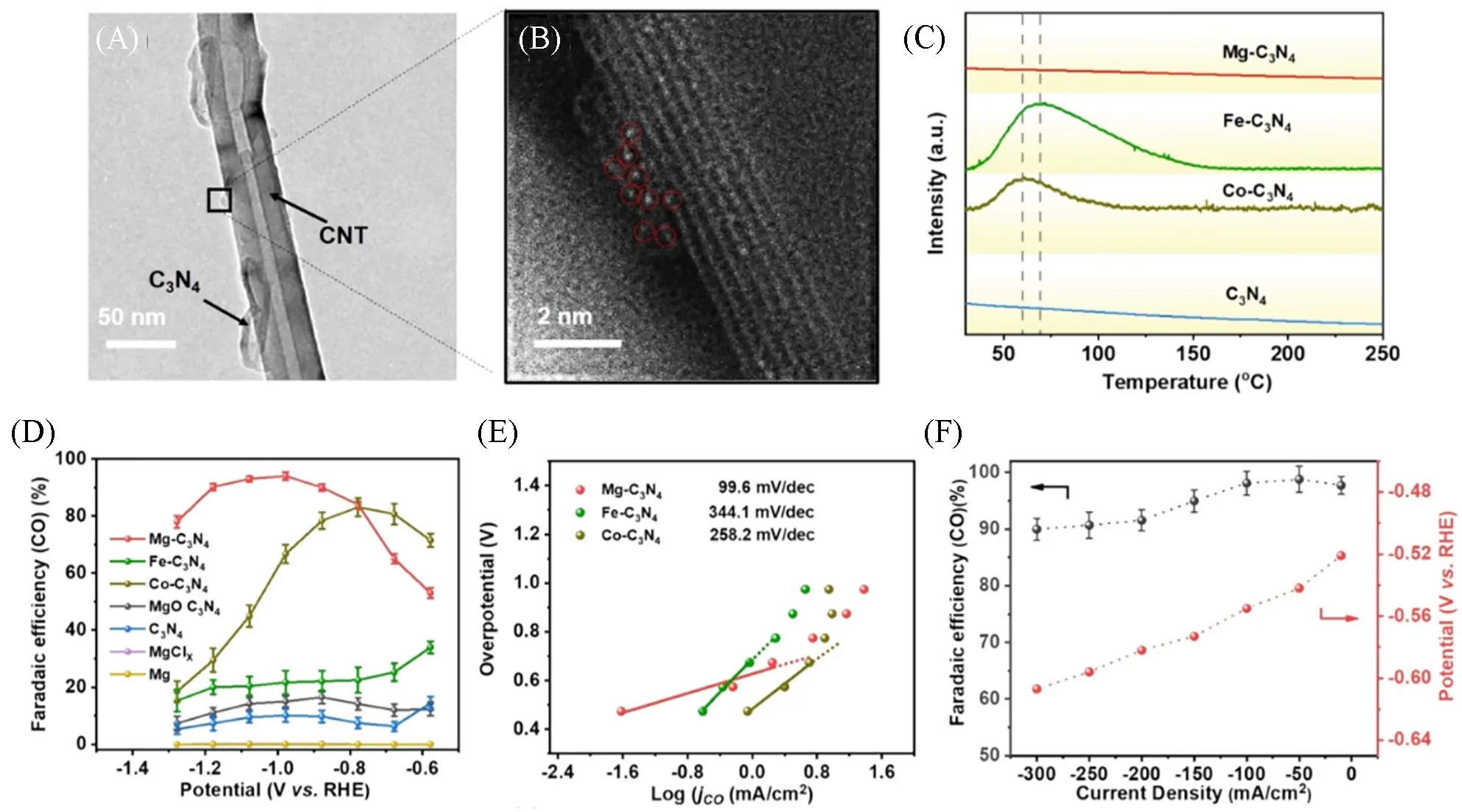
Fig.9 HRTEM(A) and AC⁃HAADF⁃STEM(B) images of Mg⁃C3N4,CO TPD curves(C),FECO at different potentials in H⁃cell(D),Tafel plots(E),potentials and FECO at different current densities in flow cell(F)[115]
值得一提的是,Cu基催化剂通常被认为是一种理想的CO2RR催化剂[122~125],目前Cu单原子催化剂也被应用于生成CO[126],CH4[127,128],CH3OH[129,130]等C1产物或者附加值更高的多碳产物.如,Zhao等[131]合成了与氮配位的Cu单原子催化剂(Cu-SA/NPC)[图10(A)],其可在较低的过电位下将CO2还原为乙醇、乙酸和丙酮,其中丙酮为主要产物.从产物分布图中可以发现,在−0.16 V 时就有含氧产物的生成.更值得注意的是,在这些还原产物中,丙酮是主要的产物.当电位处于−0.36 V时,丙酮的法拉第效率达到36.7%,远高于其它含氧产物的法拉第效率[图10(B)和(C)].在5个循环的测试中,法拉第效率几乎维持在36.7%,证明了Cu-SA/NPC具有良好的稳定性[图10(D)].理论计算结果表明,与4个吡咯氮配位的Cu是主要的活性位点,这样的配位环境有利于CO2的活化,并且降低了C⁃C偶联的自由能[图10(E)].
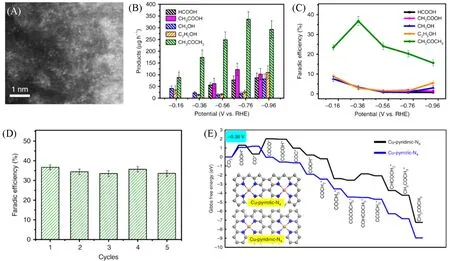
Fig.10 HAADF⁃STEM image of Cu⁃SA/NPC(A),production rate of CO2 reduction products on Cu⁃SA/NPC(B),Faradaic efficiency of CO2 reduction products on Cu⁃SA/NPC(C),stability of Cu⁃SA/NPC(D),free energy diagrams calculated at a potential of -0.36 V for CO2 reduction to CH3COCH3 on Cu⁃pyridinic⁃N4 and Cu⁃pyrrolic⁃N4 sites of Cu⁃SA/NPC(insets:the computational models)(E)[131]
3.3 电解水反应
氢气作为二次能源,因其具有清洁、能量密度高等优点,越来越受到人们的青睐.在众多制氢技术中,电解水制氢技术能够通过电能将水分解为氢气和氧气,被认为是目前最有前景的技术之一[132~136].电解水反应由阳极的析氧反应(Oxygen Evolution Reaction,OER)和阴极的析氢反应(Hydrogen Evolution Reaction,HER)构成.在实际反应中,由于浓差极化等因素的影响,往往需要施加高于理论值(1.23 V)的电压来驱动电解水反应的发生.因此,亟需开发高效的OER和HER催化剂来提升电解水的效率.Cao 等[50]报道了一种锚定在氮碳载体,具有Ru-N4位点的Ru 单原子催化剂(Ru-N-C),该催化剂可作为酸性条件下高效的OER 催化剂[图11(A)和(B)].从LSV 曲线可以看出,Ru-N-C 在0.5 mol/L H2SO4中展现出较高的催化活性[图11(C)].在电流密度为10 mA/cm2处,Ru-N-C的过电位为267 mV,质量活性和转换频率(TOF)值分别达到3571 A/gRu和3348 O2/h[图11(D)].此外,该催化剂在酸性OER 中展现出良好的稳定性,经过30 h 的运行后,活性没有发生明显的衰减[图11(E)].理论计算结果表明,O-Ru1-N4位点是该催化剂高OER 活性和稳定性的来源[图11(F)].Ir单原子作为电解水催化剂也得到了广泛的研究.Wang等[137]合成了负载于NiO上的高载量Ir单原子催化剂(Ir18%-NiO),其中Ir原子处于NiO的表层,并占据Ni原子的位置[图11(G)~(I))].从LSV曲线可知,该催化剂在1 mol/L KOH介质中展现出优异的OER活性,在10 mA/cm2处的过电位仅为215 mV,明显低于NiO和其它IrO2催化剂[图11(J)和(K)].研究表明,Ir单原子不仅可以作为催化OER的活性中心,还可以改善周围Ni原子的活性.Wang等[138]还将Ir单原子掺入NiP中来改善其OER性能.

Fig.11 HAADF⁃STEM image of Ru⁃N⁃C(A),the R⁃space curve⁃fitting of ex situ Ru⁃N⁃C(B),LSV curves of the Ru⁃N⁃C and commercial RuO2/C in 0.5 mol/L H2SO4(C),TOF and mass activities for Ru⁃N⁃C and RuO2/C(D),plot of current density and Ru dissolved mass ratio for Ru⁃N⁃C at 1.49 V in 0.5 mol/L H2SO4(E),free energy diagram for OER on Ru1⁃N4,O⁃Ru1⁃N4 and HO⁃Ru1⁃N4(F)[50],HAADF⁃STEM image of Ir single atoms(G)and corresponding atomic models(H,I),OER LSV curves of Ir⁃NiO,NiO,and IrO2 catalysts in 1 mol/L KOH(J)and corresponding overpotentials at 10 mA/cm2(K)[137]
Rh单原子也被用作掺杂剂来同时提升HER和OER的性能.Xu等[139]通过离子交换的方法,得到了生长在Cu 泡沫上的Rh 单原子掺杂的CuO 纳米阵列(Rh SAC-CuO NAs/CF)[图12(A)].通过AC-STEM可以观察到,一定数量的Rh单原子分散在CuO的晶格中,即CuO结构中的部分Cu位点被Rh单原子取代[图12(B)和(C)].性能测试结果显示,Rh单原子的掺入显著地改善了CuO的性能.在析氧反应中,当电位为1.5 V时,Rh SAC-CuO NAs/CF的电流密度可达84.5 mA/cm2,是Ir/C/CF的9.7倍[图12(D)].相比于CuO NAs/CF,Rh SAC-CuO NAs/CF 的析氢活性得到大幅提升,在10 mA/cm2处的过电位仅为44 mV,与商业Pt/C的性能相当[图12(E)].更引人注目的是,当Rh SAC-CuO NAs/CF作为双功能催化剂用于水分解时,到达10 mA/cm2所需的电压仅为1.51 V[图12(F)].密度泛函理论计算结果揭示了其高催化活性的来源.对于OER 而言,Rh SAC-CuO 对反应中间体O*具有最佳的吸附能,从而降低了反应决速步骤(OOH*→O*)的能垒;在HER中,相比于CuO,Rh SAC-CuO,其对反应中间体H*的结合能增强.同时,Rh SAC-CuO展现出更强的亲氧性,加速了水分子的解离,从而改善了反应动力学过程.除此之外,非贵金属(如In单原子)的掺杂也可以有效地提升HER 性能.Zhu等[140]在亚纳米级Pt 纳米线表面掺杂了In 单原子(SA In-Pt NWs)[图12(G)].测试结果表明,In 单原子的引入有效地增强了Pt NWs在1 mol/L KOH中的HER活性:SA In-Pt NWs达到10 mA/cm2时的过电位仅为46 mV,明显低于Pt NWs(87 mV)、Pt/C(94 mV)及其它催化剂.此外,在过电位为46 mV处,SA In-Pt NWs具有更高的质量活性,进一步证明了该策略的可行性和有效性[图12(H)和(I)].机理研究表明,超细的一维纳米结构与单原子In掺杂的结合有效地增加了活性位点数目,并且激活了Pt原子的催化能力.此外,密度泛函理论计算结果显示,原始的Pt(111)表面与HER反应中间体的结合过强,不利于H2的脱附,而In单原子的掺杂减弱了反应中间体与催化剂表面的相互作用,从而提升了HER活性.
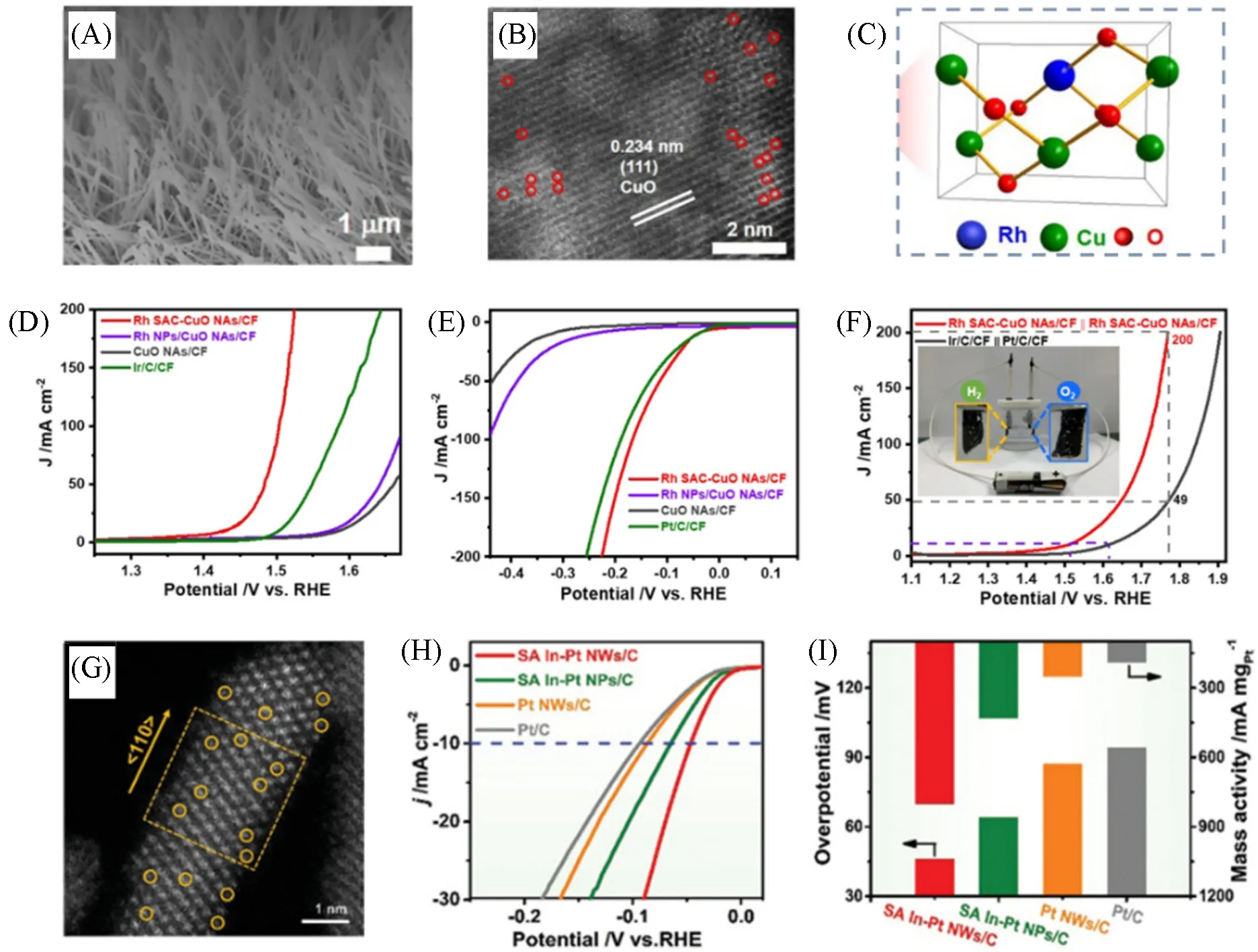
Fig.12 SEM image of Rh SAC⁃CuO NAs/CF(A),AC HAADF⁃STEM image of Rh SAC⁃CuO NAs(B),structure illustration of Rh⁃CuO(C),OER(D),HER(E)and overall water splitting performance(F)of different electrocatalysts[139],AC HAADF⁃STEM image of SA In⁃Pt NWs(G),HER performance(H)of different electrocatalysts and corresponding overpotentials and mass activities(I)[140]
3.4 氮气电还原反应
氨是一种重要的化工原料,固氮是人类生活生产中一个至关重要的化学过程[141,142].目前,工业上90%的氨都是通过传统的哈伯工艺法制取.该工艺不仅需要在高温条件下进行,而且需要消耗大量的化石燃料,对环境和能源造成较大的压力.电化学还原氮气产氨(Nitrogen Reduction Reaction,NRR)因其反应条件温和、无污染等优点,近年来受到了研究者们的广泛关注[143].然而,非极性氮氮三键的断裂仍然需要较高的能量[144].因此,开发高活性与高选择性的NRR 催化剂是推进该技术的关键之一.Tao等[145]在氮掺杂多孔碳中锚定了单原子Ru,并在载体中加入了ZrO2(Ru@ZrO2/NC)[图13(A)],用以高效的NRR.相比于Ru/C,Ru/NC,Ru@ZrO2/C 等催化剂,Ru@ZrO2/NC 展现出了更高活性,在−1.1 V处,其法拉第效率可达约21%[图13(B)].此外,Ru@ZrO2/NC具有更高的NH3分电流密度[图13(C)].值得一提的是,经过60 h的稳定性测试,Ru@ZrO2/NC的NH3产率和法拉第效率并未发生明显的衰减,证明了Ru 单原子与载体间的协同作用在NRR 中的优越性[图13(D)].理论计算进一步揭示了Ru@ZrO2/NC高性能的来源.结果表明,ZrO2表面的O空位可以锚定Ru单原子,该Ru位点对于第一步生成*NNH所需的自由能较低,从而有效地提升了催化活性[图13(E)和(F)].Li等[146]报道了一种单原子Fe 锚定在氮氧(N,O)共掺杂的反蛋白石结构的多孔纳米碳催化剂(FeSA-NO-C),Fe 中心由两个O 原子和两个N 原子共同锚定在碳基底上[图13(G)].性能测试结果表明,在900 ℃下进行碳化的FeSA-NO-C(FeSA-NO-C-900)展现出优越的NRR 活性与选择性.在−0.4 V 处,法拉第效率达到11.8%,NH3的产率为31.9明显优于其它催化剂[图13(H)和(I)].实验与理论计算结果共同证明了O物种的引入有效地调控了Fe位点的电荷分布,从而提升了FeSA-NO-C-900的反应动力学.

Fig.13 HAADF⁃STEM image of Ru@ZrO2/NC after tuning color contrast(A),FEs(B) and partial cur⁃rent densities of NH3(C) over Ru@NC,Ru@C,Ru@ZrO2/NC and Ru@ZrO2/C,the long⁃term durability test at 0.21 V over Ru@ZrO2/NC at ca. 10 ℃(D),results of DFT calculation(E,F)[145],atomic structure model of FeSA⁃NO⁃C(G),FEs(H) and NH3 yield rates(I) of FeSA⁃NO⁃C⁃800,FeSA⁃NO⁃C⁃900,FeSA⁃NO⁃C⁃1000 and FeSA⁃NO⁃C⁃900 without SiO2[146]
4 总结与展望
开发高效、低成本的催化剂对于能源转化和储能应用具有重要意义.单原子催化剂作为一类新型催化剂,由于其原子级分散的金属中心具有独特的结构和电子性质,因而在一系列电化学反应中具有替代或补充传统纳米结构催化剂的潜力.本文例举了一些单原子催化剂常用的表征手段,总结了一些常见的单原子催化剂的合成方法.随后,对单原子催化剂在各种能源转化相关的电催化反应(如ORR,CO2RR,HER,OER和NRR)中的研究进展进行了综合评述(图14).尽管目前单原子催化已经取得了令人瞩目的成绩,但在合成方法、催化剂稳定性、表征手段以及理论与实验相结合等方面仍有许多挑战有待解决.

Fig.14 Challenges and perspectives in the field of single⁃atom electrocatalysis
目前,单原子催化剂的负载量普遍偏低,且大部分单原子的合成方法仅局限于几种金属元素,缺乏广泛的普适性.而在实际应用中,催化剂的高性能需要高密度的金属单原子来获得丰富的活性位点.因此,开发简便、可规模化制备、高负载量,且经济可行的新型合成路线是目前的研究重点.
稳定性是单原子催化在实际应用中的另一个关键因素.在实际催化过程中,单原子中心的失活与碳载体的降解都会导致单原子催化剂的结构被破坏,从而造成催化剂失活.基于此,研究具有抗腐蚀性的新型单原子催化剂与具备高导电性和耐腐蚀性能的先进载体材料是目前迫切需要的.
催化剂的原子结构与电子结构共同决定了其内在的活性与催化机理.然而,明确识别单原子催化剂的原子结构,揭示其催化活性的来源以及真实催化反应过程中结构变化仍是一个严峻的挑战.为此,利用更强大的原位表征技术,以在更高的空间与时间分辨率上了解单原子的结构特点是必不可少的.原位X射线技术、原位透射电子显微镜以及原位拉曼光谱等能够提供实际反应过程中原子的电子结构、配位环境及其变化的信息,对于进一步理解真实反应过程中单原子的结构演变具有十分重要的意义.
计算化学已成为预测/解释催化剂活性的重要手段之一.相比于传统催化剂,单原子催化剂具备更加明确的活性中心.因此,在模型构建上具有更高的准确性.此外,鉴于单原子催化剂的结构在实际催化反应可能存在动态演化行为,利用原位表征结果来辅助理论计算,可以更加精准地揭示单原子催化的本质.
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中南民族大学学报(自然科学版)(2021年1期)2021-02-02
陶瓷学报(2020年2期)2020-10-27
陶瓷学报(2020年2期)2020-10-27
天津医科大学学报(2019年3期)2019-08-13
中国资源综合利用(2017年4期)2018-01-22
南方文学(2016年4期)2016-06-12
海峡科技与产业(2016年3期)2016-05-17
