
氨丙基低聚硅倍半氧烷及其酰胺化产物的合成与表征
2022-08-25 08:04董佑邦张泽祺杨荣杰
中国塑料 2022年8期
董佑邦,张泽祺,2,杨荣杰,2*
(1.北京理工大学材料学院,国家阻燃材料工程技术研究中心,北京 100081;2.北京理工大学,中原阻燃材料研究中心,河南 许昌 461000)
0 前言
多面体低聚硅倍半氧烷(POSS)是1种特殊的有机/无机纳米杂化分子,在三维结构上以Si—O—Si键构成主体分子骨架,外围则可以设计为不同的取代基,具有良好的阻燃性、耐热性及突出的抗热氧化性等优良特性[1‐3]。经过人工分子设计的反应性POSS更可以在过渡金属的配位[4‐5]、液晶材料[6]、吸附材料[7]等方面发挥多样化的功能。常见的规整笼形POSS及特殊结构的POSS都可以通过水解缩合法得到[8‐10];而对于含有特殊官能团的反应性POSS除了水解法外,也可以借助官能团之间的转化反应,常用对八苯基硅倍半氧烷(OPS)进行硝化、还原等过程来合成NO2‐POSS、NH2‐POSS[11‐12]。氨基 POSS 取代基上的氨基具有不错的反应活性,既可以作为大分子原料进行分子设计,又在高分子树脂基材料、气凝胶材料、功能性薄膜等应用中具有良好的相容性[13‐17]。Fan 等[18]利用八氨基苯基POSS(OAPS)与溴丙炔反应,成功合成了1种新型的八炔丙基POSS。作为原料的OAPS合成需要经历苯基硝化、硝基还原的过程,合成周期长,产物的官能度较难把控,因此使用带氨基的硅烷进行水解缩合是另一种合成氨基POSS的常用方法。通常水解法使用浓盐酸催化可以得到八官能度氨基 POSS[19‐20],反应时间在72 h至7 d不等。带有氨基的硅烷在反应体系呈较强的碱性,有报道称浓盐酸长时间催化容易与之结合形成氨基POSS的盐酸盐[21],而使用无机强碱(如KOH[22])在起到催化作用的同时可以避免该现象的发生。用碱催化代替浓盐酸催化氨基硅烷的水解缩合是1种有效且不会影响产物结构的合成方法。
本文以 NH2‐propyl‐silane为原料,使用碱催化水解缩合一锅法合成了不同Si原子数的NH2‐propyl‐POSS,反应时间较短,并利用氨基进行酰胺化合成了2种新型酰胺化笼形硅倍半氧烷;采用红外光谱仪(FTIR)、核磁共振分析仪(NMR)、质谱仪(MS)、X射线粉末衍射仪(XRD)和热重分析仪(TG)对产物的化学结构和热稳定性进行了详细的表征;制备了EP/NH2‐propyl‐POSS,并通过TG、差示扫描量热仪(DSC)和氧指数测试仪对其性能进行了测试。
1 实验部分
1.1 主要原料
NH2‐propyl‐silane,纯度 97%,上海迈瑞尔化学技术有限公司;
氢氧化钾、无水硫酸镁、丙酮、乙酸、无水乙醇,分析纯,北京市通广精细化工公司;
EP,E‐44,肥城德源化工有限公司;
二氨基二苯砜(DDS),纯度97%,北京华威锐科化工有限公司;
盐酸,含HCl 36%,北京市通广精细化工公司;
丙炔酸,纯度95%,安徽泽升科技有限公司。
1.2 主要设备及仪器
FTIR,6700,美国Nicolet公司;
NMR,Avance600(600 MHz),瑞士Bruker公司;
MS,micrOTOF‐Q Ⅱ,ESI离子源,瑞士Bruker公司;
XRD,Rigaku D/MAX 2500,日本理光公司;
TG,209F1,德国NETZSCH公司;
DSC,204F1,德国NETZSCH公司;
氧指数测试仪,FTA II,英国RS公司;
鼓风干燥箱,DF206,上海一恒科学仪器有限公司;
电磁搅拌锅,DF101S,北京星德仪器设备有限公司。
1.3 样品制备
NH2‐propyl‐POSS合成:采用一锅法合成,在三口烧瓶中加入50 mL丙酮,称取0.1 g KOH溶于5 mL去离子水配成碱液,将两者混合搅拌均匀;称取9.22 g NH2‐propyl‐silane(约 0.05 mol)置于低于 10 ℃冰水浴条件,用恒压滴液漏斗将其滴入三口瓶中的反应溶液,10~15 min滴完,持续搅拌20 min;然后将盛有反应溶液的烧瓶转移到油浴锅中,控制温度在65~70℃进行缩合反应24 h;反应结束后滴加少量盐酸中和反应溶液中剩余的KOH,经过滤、旋蒸除去丙酮与水,得到固体产物在乙醇中再次溶解,并用无水硫酸钠除水,再次旋蒸除去乙醇,得到褐黄色粉末,在60℃下烘干3 h得到6 g产物,产率为60%;
CH3‐CONH‐POSS合成:延续采用一锅法合成,在水解缩合反应后未经后处理的NH2‐propyl‐POSS反应溶液中滴加入乙酸,有油状液体在下层析出,滴加至反应液pH值为7后持续搅拌1 h,收集底部的油状液体,用乙醇多次溶解、除水、旋蒸,得到浅棕色固体粉状物,在60℃下烘干3 h;
CH≡C—CONH‐POSS合成:延续采用一锅法合成,在未经后处理的NH2‐propyl‐POSS反应溶液中滴加入丙炔酸,其余步骤同CH3‐CONH‐POSS,最终得到浅褐色粉末固体,在60℃下烘干3 h;
EP/NH2‐propyl‐POSS共混物制备:采用热固化工艺,在140 ℃下将合成的NH2‐propyl‐POSS通过机械搅拌在E‐44中分散均匀;随后加入适量的固化剂DDS,继续搅拌15~30 min后倒入聚四氟乙烯模具中,在180℃下热固化4 h;固化结束后脱模得到EP/NH2‐pro‐pyl‐POSS共混物;共混物中各组分含量如表1所示。
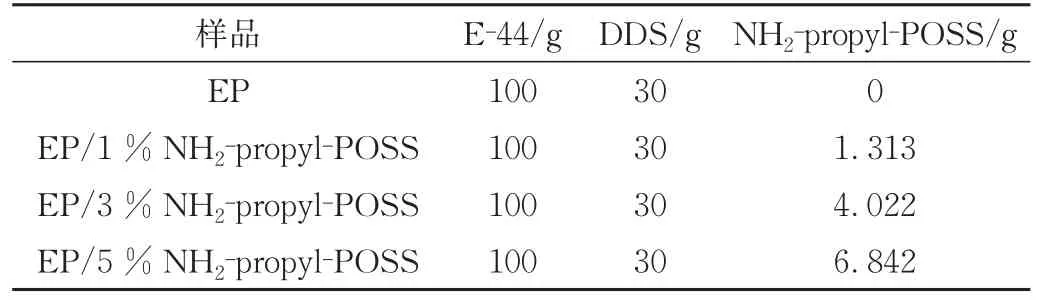
表1 EP/NH2⁃propyl⁃POSS共混物各组分含量Tab.1 Content of each component of EP/NH2‐propyl‐POSS blends
1.4 性能测试与结构表征
红外分析:分辨率为4 cm-1,扫描次数为32,扫描范围为4 000~400 cm-1;
核磁共振分析:1H‐NMR、13C‐NMR和29Si‐NMR所用溶剂为D2O,扫描次数分别为32、128、512;
质谱分析:样品溶于甲醇配成0.1‰溶液,过滤后注入MS,记录数据;
X射线衍射分析:样品研磨过200目筛,测试范围为2°~90°,扫描速率为6(°)/min;
热重分析:将3种POSS样品在氮气气氛下,以10℃/min的速率从40℃升温到900℃;共混物在氮气气氛下,以20℃/min的速率从40℃升温到800℃,考察其热失重情况;
热性能分析:将样品在氮气气氛下,以20℃/min的速率从40℃升温到300℃,恒温1 min后以20℃/min的速率降温至100℃;二次升温以10℃/min的速率从100℃升温到300℃;
LOI测试:浇注尺寸为100 mm×6.5 mm×3 mm,根据GB/T 2406.1—2008进行测试,每组测试5根。
2 结果与讨论
2.1 NH2⁃propyl⁃POSS的合成与表征
NH2‐propyl‐POSS 通过 NH2‐propyl‐silane 在碱性环境下的水解缩合反应得到,以含有8个Si原子的T8笼型结构为例,反应过程如图1所示。得到的NH2‐pro‐pyl‐POSS在一些常见试剂中的溶解性如表2所示。
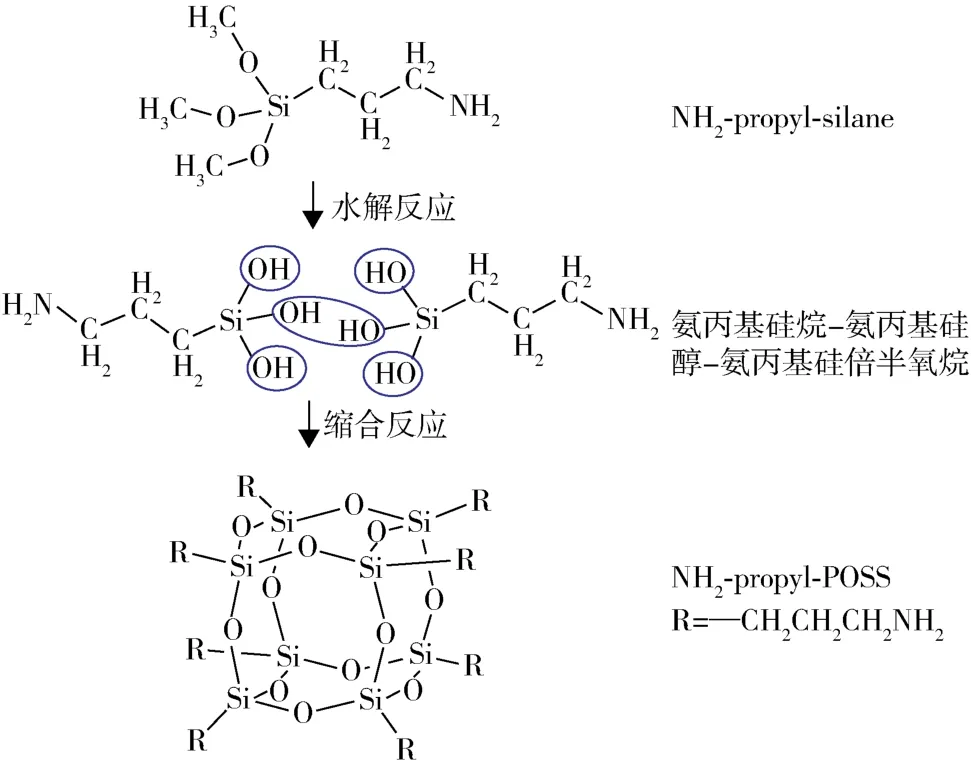
图 1 NH2‐propyl‐POSS 的合成步骤Fig.1 Synthesis process of NH2‐propyl‐POSS

表2 室温下NH2⁃propyl⁃POSS在常用溶剂中的溶解性Tab.2 Solubility of NH2‐propyl‐POSS in common solvents at room temperature
图 2 为 NH2‐propyl‐silane和 NH2‐propyl‐POSS 的FTIR谱图。图中NH2‐propyl‐silane在1 100、2 840 cm-1处的尖锐吸收峰分别是Si—O—C和—OCH3的特征吸收峰;3 363、1 587 cm-1处较弱的吸收峰分别是伯胺基团的N—H的伸缩振动吸收峰和弯曲振动吸收峰[18];在经过水解缩合反应后,2 840 cm-1处—OCH3的特征吸收峰明显消失,同时1 000~1 100 cm-1处出现了明显的Si—O—Si特征双吸收峰,初步说明原料中的—OCH3已经水解完全及Si—O—Si结构的形成;在3 200~3 500 cm-1范围内存在多处鼓包峰,3 365 cm-1处为氨基N—H的伸缩振动吸收峰,3 271 cm-1处应是Si—OH中—OH的吸收峰。

图2 样品的FTIR谱图Fig.2 FTIR spectra of the samples
图 3(a)是原料 NH2‐propyl‐silane 和产物 NH2‐pro‐pyl‐POSS的1H‐NMR谱图,氘代试剂为重水。在原料中,0.53、1.59、2.74处的3处信号峰为丙基上亚甲基氢(—CH2—)的信号峰;3.26处信号峰归属为甲氧基(—OCH3)上的氢,4.71处为溶剂水峰。在氢谱中没有检测到明显的氨基(—NH2)信号峰,这是因为—NH2上活泼氢在溶剂中交换太快[23]。产物 NH2‐propyl‐POSS中,—OCH3的氢信号峰已完全消失。由图3(b)13C‐NMR 谱图可以看出,NH2‐propyl‐silane在 10.30、23.26、42.66处有3处亚甲基的碳原子峰,48.84处为—OCH3中甲氧基的碳原子峰,该信号峰在NH2‐propyl‐POSS的碳谱中完全消失,氢谱和碳谱结果证明了活性基团—OCH3的完全水解。

图3 样品的NMR谱图Fig.3 NMR spectra of the samples
图 3(c)、(d)分别为原料 NH2‐propyl‐silane和产物NH2‐propyl‐POSS 的29Si‐NMR 谱图。可以看出 NH2‐propyl‐silane在-42.01和-50.34处有 2个硅原子峰,-42.01处的硅原子峰为氨丙基三甲氧基硅烷中的硅原子信号峰,-50.34处硅原子峰的出现可能是因为硅烷二聚体的形成[24]。NH2‐propyl‐POSS 在-57.44和-58.90处新出现了2个鼓包状信号峰,其中-58.90处为完全缩合的Si—O—Si笼形结构中的硅原子信号峰,-57.44处为部分没有完全缩合的带有—OH的硅原子信号峰[25]。
从NMR谱图可以看出NH2‐propyl‐silane水解反应完全,但是NH2‐propyl‐POSS中存在一部分未完全缩合Si—OH。
图4为产物 NH2‐propyl‐POSS的ESI‐TOF谱图。可以看出 NH2‐propyl‐POSS 的m/z信号峰分布在500~1 500之间,不同m/z值的信号峰对应不同Si原子数的NH2‐propyl‐POSS。表3给出了通过计算得到的不同m/z值所对应的NH2‐propyl‐POSS 的大概结构。可以看出,m/z值为661.2、881.2、1 101.3和1 323.4代表完全缩合的笼形/环梯形T6、T8、T10和T12结构;m/z值为763.2、863.2、1 000.3和1 221.3则代表含有Si—OH的不完全缩合的多角笼形T7、T8、T9和T11结构。在不完全缩合结构中,注意到部分结构的实际分子量与m/z值有所不符,以T9结构为例,计算分子量为999,而m/z值在1 000.3的信号峰左侧还存在m/z值为982.3的信号峰,m/z值982.3与计算分子量999的差值为17,正好是1个—OH的质量数;同样在T8结构中,带有Si—OH的不完全缩合结构计算分子量为898,实际m/z值为863.2,与计算分子量之间的差值为34,是2个—OH的质量数。类似的现象在张文超合成含磷POSS的质谱中也有发生[24],出现这一现象的原因可能是一部分不完全缩合的NH2‐propyl‐POSS在检测过程中发生了—OH的掉落。
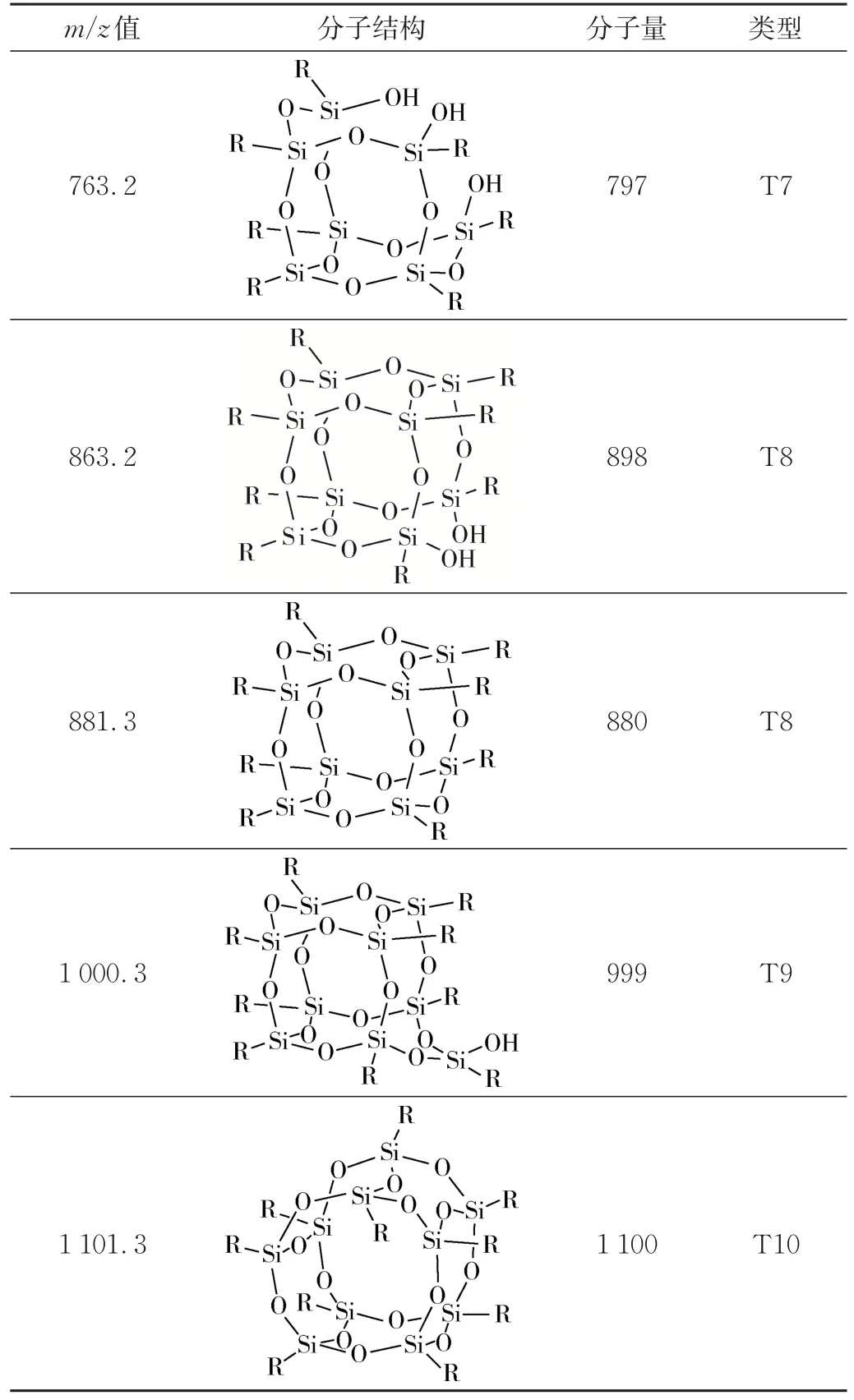
表3 NH2⁃propyl⁃POSS的结构Tab.3 Structures of NH2‐propyl‐POSSs
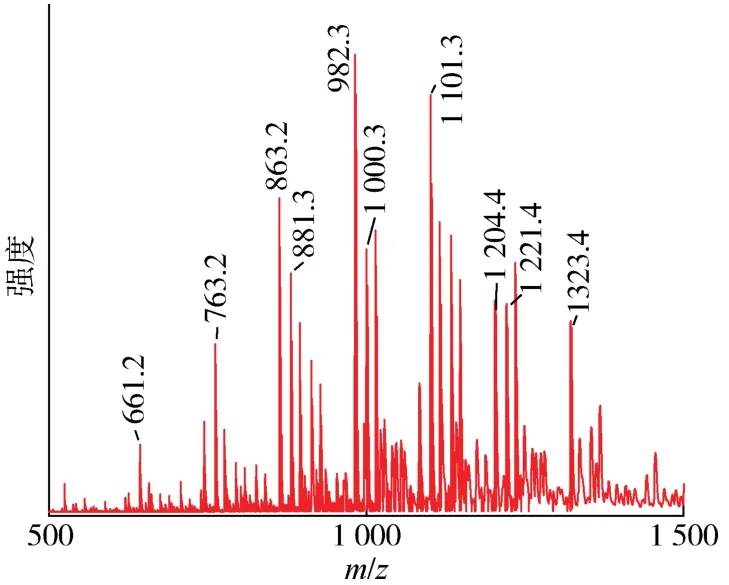
图 4 NH2‐propyl‐POSS 的 ESI‐TOF 谱图Fig.4 ESI‐TOF mass spectrum of NH2‐propyl‐POSS
从信号峰的强度与m/z值可以看出m/z值的分布符合一定的正态分布,产物NH2‐propyl‐POSS中信号较强的是T8、T9和T10结构。质谱解析的结果与FT‐IR谱图、NMR谱图都有比较好的吻合,进一步说明合成了一系列不同Si原子数的NH2‐propyl‐POSS产物,包括完整笼形的T6、T8、T10、T12完全缩合POSS以及带Si—OH的不完全缩合POSS。
2.2 酰胺化POSS的合成与表征
NH2‐propyl‐POSS中的伯氨基(—NH2)具有很高的反应活性。缩合反应结束后,在NH2‐propyl‐POSS反应液中滴加有机羧酸,既可以起到中和剩余KOH的作用,又可以与NH2‐propyl‐POSS取代基上的—NH2发生进一步酰胺化反应。本文使用乙酸和丙炔酸,利用羧酸基团与氨基的酰胺化反应,得到了2种NH2‐pro‐pyl‐POSS的酰胺化衍生物,分别为CH3‐CONH‐POSS和CH≡C—CONH‐POSS。表4给出了2种酰胺化POSS在一些常见溶剂中的溶解性。

表4 CH3⁃CONH⁃POSS和CH≡C—CONH⁃POSS的溶解性Tab.4 Solubility of CH3‐CONH‐POSS and CH≡C—CONH‐POSS
图 5 为 NH2‐propyl‐POSS、CH3‐CONH‐POSS 和CH≡C—CONH‐POSS的FTIR谱图。在CH3‐CONH‐POSS和CH≡C—CONH‐POSS的谱图中,新出现1 300~1 600 cm-1处的双峰,其中1 388 cm-1处吸收峰为仲氨基N—H弯曲振动和C—N伸缩振动的1个叠加吸收峰,1 530 cm-1为C=O伸缩振动峰,这处双峰的出现也证明—NH2与—COOH发生酰胺化形成了—NHCO—基团。除此之外,在CH≡C—CONH‐POSS的谱图中还可以看到2 081 cm-1处C≡C的伸缩振动吸收峰,685 cm-1处炔基氢(—C≡CH)的弯曲振动吸收峰,没有检测到明显的炔氢伸缩振动峰。
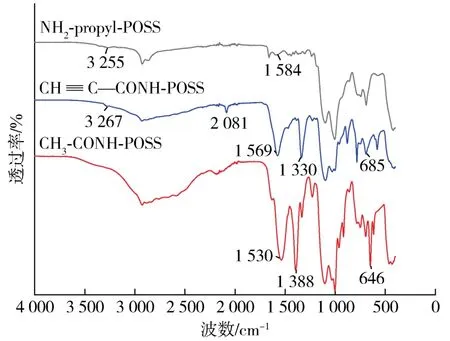
图5 3种POSS的FTIR谱图Fig.5 FTIR spectra of the three POSSs
图 6(a)给出了 NH2‐propyl‐POSS 和 2 种酰胺化POSS的1H‐NMR谱图,其中4.71处为D2O水峰。可以看出2种酰胺化POSS均在0.63、1.69、2.93处出现—CH2CH2CH2NH2中的3处亚甲基氢原子峰。在CH3‐CONH‐POSS的1H‐NMR谱中,1.75处氢原子峰为乙酰基(—NHCOCH3)上的甲基氢原子峰。由于D2O活泼氢交换作用的影响[23],没有检测到—CONH—中仲氢(—NH—)原子的信号峰。
从图6(b)中可以看出,在CH≡C—CONH‐POSS的13C‐NMR谱图中,除9.17、20.81、42.68 3处亚甲基的碳原子峰外,70.65和78.66出现2处明显的—C≡CH中炔基碳原子峰,同时C=O中羰基碳原子峰出现在160.01处。CH3‐CONH‐POSS的13C‐NMR 谱图中23.31处新出现了羰甲基上甲基的碳原子峰,羰基的碳原子峰出现在181.37处。13C‐NMR谱图的表征结果很好地证明了 NH2‐propyl‐POSS上的—NH2与有机羧酸中的—COOH发生了酰胺化反应。
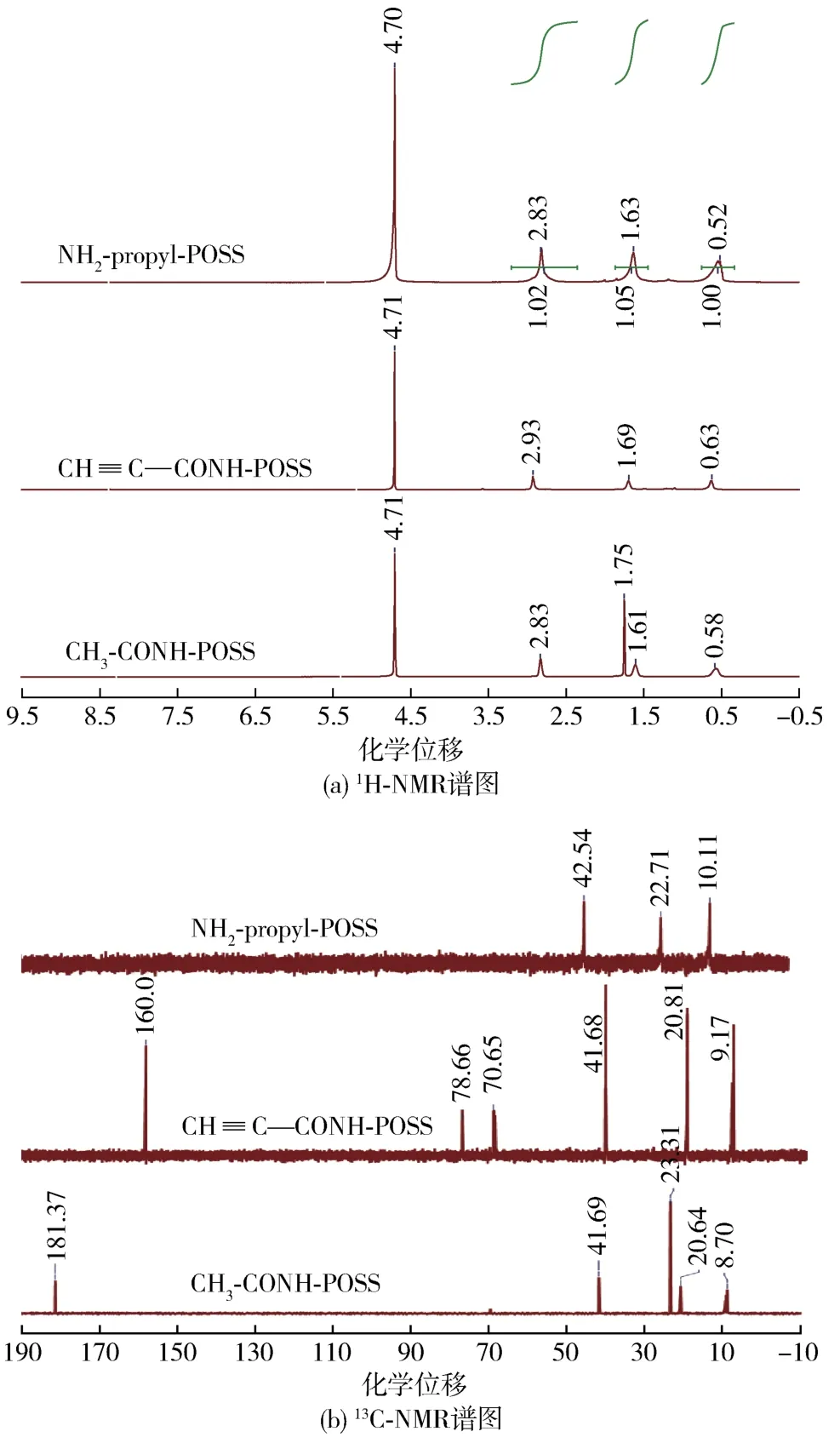
图6 3种POSS的核磁共振谱图Fig.6 NMR spectra of the three POSSs
2.3 XRD分析
图 7为NH2‐propyl‐POSS和2种酰胺化POSS的XRD谱图。从图中可以看出3种POSS均为无定形态。NH2‐propyl‐POSS在2θ为6.56°、21.34°有2处衍射峰,其中小角区6.56°处的尖锐峰与NH2‐propyl‐POSS的分子主链间距有关,21.34°处的宽峰则是取决于POSS笼的分子结构[12]。CH3‐CONH‐POSS 和 CH≡C—CONH‐POSS的XRD谱图也说明酰胺化的过程中,NH2‐propyl‐POSS的基本笼形结构得到保留,仅在POSS的取代基上产生了—CONH—基团。上述变化反映在11.54°处新出现1处鼓包状衍射峰,这可能是—CONH—基团的存在造成了POSS上的取代基链在空间堆积分布上的变化。

图7 3种POSS的XRD谱图Fig.7 XRD patterns of the three POSSs
2.4 TG分析
3种POSS的TG和DTG曲线如图8所示,相关的温度数据在表5中给出。从图表中可以看到NH2‐propyl‐POSS在氮气气氛下的TG曲线呈二级阶梯状,DTG曲线上有2处峰值,说明NH2‐propyl‐POSS的失重过程有2个主要阶段。在NH2‐propyl‐POSS的失重过程中,失重5%的温度(Tonset)为274.6℃,900℃下的残余质量为44.15%,2个阶段所对应的最大失重速率温度(Tmax1、Tmax2)分别为331.1℃和470.3 ℃,表明NH2‐propyl‐POSS具有良好的热稳定性。从DTG曲线上可以观察到,NH2‐propyl‐POSS在200~300℃范围内失重非常缓慢,结合质谱解析的结构来看,在此温度段内的失重可能是未完全缩合的Si—OH之间脱水导致的;在温度上升到300~600℃之后,NH2‐propyl‐POSS进入快速失重的阶段,在331.1 ℃下的失重速率达到-3.10%/min,470.3℃下的失重速率达到-1.94%/min,这部分失重的主要原因是Si—O—Si笼形结构和侧链取代基3‐氨基丙基的分解和重组。

表5 3种POSS的热稳定性Tab.5 Thermal stability of the three POSSs

图8 3种POSS的TG和DTG曲线Fig.8 TG and DTG curves of the three POSSs
NH2‐propyl‐POSS为Si—O—Si笼型结构,每个顶点Si原子都带有3‐氨基丙基取代基,在300~600℃失重过程中有2个明显阶段,说明氨基、丙基及Si—O—Si笼形结构的热分解存在先后顺序。一般Si—O键键能为443 kJ/mol,—NH2作为 NH2‐propyl‐POSS 的端基,非共轭C—N键的键能为332 kJ/mol,小于C—C键的键能368 kJ/mol,在受热情况下,C—N键相比于C—C键更容易断键。因此300~400℃范围内的失重的主要原因可能是:(1)—NH2的脱落;(2)与—NH2相连的部分—CH2—的断键。而400~600℃范围内失重的主要原因则是丙基碳链的分解以及Si—O—Si笼形结构破坏。以完全缩合的T8结构NH2‐propyl‐POSS为例,其带有8个氨基丙基取代基,分子量为880.3,分子中所有有机基团的分子量总和为464。当有机基团全部分解之后,剩余的Si—O化合物占整个大分子质量的理论比例约为47.27%,与测试结果的44.15%十分接近。
经过酰胺化的CH≡C—CONH‐POSS和CH3‐CONH‐POSS的TG曲线也呈现台阶状,失重过程中也有2个主要阶段,但是失重过程及其对应的温度相比NH2‐propyl‐POSS有较大的变化。从曲线来看,2种酰胺化POSS的Tonset都有很大的降低,CH3‐CONH‐POSS为144.3℃,CH≡C—CONH‐POSS为120.8℃。2种酰胺化POSS的第一个失重阶段基本在300℃前结束,而NH2‐propyl‐POSS在该温度下才刚刚开始进行第一个阶段的失重。CH3—CONH‐POSS在177.9℃达到Tmax1,失重第一阶段在200℃结束,在此阶段失重达到26.81%。CH≡C—CONH‐POSS在124.9℃达到Tmax1,第一阶段在150℃结束,在此阶段失重15.92%。254~600℃是2种酰胺化POSS的第二个失重阶段,Tmax2均在435℃以上,相比NH2‐propyl‐POSS提前了30℃。900℃下CH≡C—CONH‐POSS和CH3‐CONH‐POSS的残余质量分别为41.86%和41.74%,应归于POSS笼形Si—O—Si结构产生的氧化硅。
2.5 EP/NH2⁃propyl⁃POSS共混物的热性能
通过TG研究了纯EP 以及EP/NH2‐propyl‐POSS共混物在氮气气氛下的热稳定性,测试结果见图9和表6。在氮气气氛下,EP/NH2‐propyl‐POSS 共混物与纯 EP相比,热分解过程没有明显变化。添加NH2‐propyl‐POSS 后,EP/NH2‐propyl‐POSS 共混物的Tonset和Tmax稍有下降,而NH2‐propyl‐POSS 的Tonset远低于纯 EP,这说明NH2‐propyl‐POSS在EP基体中没有发生单独的热分解,而是在升温过程中与EP基体发生作用,进而使得基体的热分解有所提前。此外EP/NH2‐propyl‐POSS 共混物的Tmax随着NH2‐propyl‐POSS含量的逐渐增加呈现降低趋势,而800℃下的残余质量却随着NH2‐propyl‐POSS含量的逐渐增加而增加。

图9 EP/NH2‐propyl‐POSS共混物的TG和DTG曲线Fig.9 TG and DTG curves of EP/NH2‐propyl‐POSS blends

表6 EP/NH2⁃propyl⁃POSS共混物的热稳定性Tab.6 Thermal stability of EP/NH2‐propyl‐POSS blends
EP/NH2‐propyl‐POSS共混物的Tg通过DSC测试得到,从图10可以看出,不同EP/NH2‐propyl‐POSS共混物都只有1个Tg,这说明NH2‐propyl‐POSS和EP基体有较好的相容性。随着NH2‐propyl‐POSS含量的增加,EP/NH2‐propyl‐POSS 共混物的Tg逐渐升高,在 NH2‐propyl‐POSS含量为3%时涨幅最大,原因是在热固化过程中一部分NH2‐propyl‐POSS取代基上的—NH2能够参与环氧基开环,以刚性侧链基团或者交联点的形式嵌入环氧树脂固化后的交联网络,从而提高EP/NH2‐propyl‐POSS共混物的Tg。

图10 EP/NH2‐propyl‐POSS共混物的DSC曲线Fig.10 DSC curves of EP/NH2‐propyl‐POSS blends
2.6 EP/NH2⁃propyl⁃POSS共混物的LOI
EP/NH2‐propyl‐POSS共混物的LOI由表7列出。随着 EP/NH2‐propyl‐POSS共混物中NH2‐propyl‐POSS含量的增加,LOI逐渐增加;从纯EP的22.0%依次提高到22.4%、24.8%、25.2%。从图11可以看出,未添加NH2‐propyl‐POSS前,纯EP样条经LOI测试后的残炭所剩无几;添加 3%NH2‐propyl‐POSS 后的样条测试后形成了大量残炭,这是因为NH2‐propyl‐POSS的添加能够提高EP基体燃烧后的残炭率。

表7 EP/NH2⁃propyl⁃POSS共混物的LOITab.7 LOI of EP/NH2‐propyl‐POSS blends

图11 LOI测试后的样条Fig.11 Splines after LOI test
3 结论
(1)由NH2‐propyl‐silane通过碱催化水解缩合一锅法合成的NH2‐propyl‐POSS是完全缩合POSS和不完全缩合POSS混合物,其中含量较高的是T8、T9、T10结构的POSS;采用一锅法通过乙酸和丙炔酸与NH2‐propyl‐POSS 进行酰胺化反应,合成了 CH3‐CONH‐POSS和CH≡C—CONH‐POSS,FTIR、1H‐NMR、13C‐NMR分析结果证明—NH2与—COOH发生了酰胺化反应,形成了酰胺化POSS;
(2)NH2‐propyl‐POSS 的Tonset为 274.6 ℃,Tmax2可达470.3℃,具有很好的热稳定性;CH≡C—CONH‐POSS和CH3‐CONH‐POSS的Tonset分别为144.3 ℃和128.8℃,Tmax2均在435℃以上;3种POSS在900℃下的残余质量均在40%以上,是源于POSS氧化硅残余物;
(3)在EP基体中添加NH2‐propyl‐POSS可以提高基体的热稳定性及Tg,当 NH2‐propyl‐POSS 含量达到5% 时 ,EP/NH2‐propyl‐POSS 共 混 物 的Tonset为378.8℃,残余质量达到14.23%,Tg提高至180.7℃,LOI提高至25.2%。
猜你喜欢
农药学学报(2022年3期)2022-06-14
节能与环保(2022年3期)2022-04-26
安徽农学通报(2022年6期)2022-04-07
林业工程学报(2022年1期)2022-02-26
看世界·学术下半月(2020年5期)2020-09-10
当代化工(2020年8期)2020-09-09
当代水产(2020年3期)2020-06-15
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
