
基于木香药材-标准汤剂-配方颗粒的指纹图谱及含量测定研究
2022-07-27 02:58韩慧琴狄慧荆燕燕杨桂英尹贻珍
世界最新医学信息文摘 2022年24期
韩慧琴,狄慧,荆燕燕,杨桂英,尹贻珍
(张家口市食品药品检验中心,河北 张家口 075000)
0 引言
木香(Aucklandiae Radix )来源于菊科植物木香的干燥根。木香可以行气止痛、调中导滞,如果患者有脾胃气滞,脘腹胀满或者出现疼痛,并且伴有嗳气、恶心、呕吐的患者,可以应用木香进行治疗[1]。木香在临床上是比较常用的一种中药材,古代医家将其列为上品[2]。其主要含有萜类,蒽醌类,生物碱类,黄酮类等。目前关于木香的研究多为含量测定[3]、药理作用[4]和药材的指纹图谱[5]等方面。关于木香配方颗粒的报道较少。
中药配方颗粒是通过现代化的制药手段,以中药饮片为初始原料,用水煎煮提取、放冷过滤、减压干燥、制粒成形,其药效物质、性味归经、功能主治和传统中药汤剂一致,既能保证中医辨证论治的特点、又可以灵活加减,方便患者服用,卫生有效[6-8]。国家药监局发布2021年第22 号公告,结束中药配方颗粒试点工作,说明配方颗粒具备很多优点,并且已被接受,故国家鼓励生产配方颗粒。但是从质量标准来看,目前检验项目还不是很全面,特别是安全性有效性方面还有待提高。因此,课题组参考有关文献[9,10]采用HPLC 法建立了指纹图谱及含量测定方法,可为木香配方颗粒的质量控制提供参考依据,可为其真伪优劣鉴别提供参考手段。
1 仪器与试药
LC-30AT 型高效液相色谱测定仪(配备二极管阵列检测器)(日本岛津公司); e2695-2998(配备二极管阵列检测器)型高效液相色谱仪(美国Waters 公司);国家药典会“中药指纹图谱相似度评价软件(版本号为2012.130723)”;统计软件(SPSS 19.0);BT125D 型精密十万分之一电子天平(德国sartorius,分度:0.01mg);DTC-27 型静音型超声波清洗机(湖北鼎泰高科有限公司);木香烃内酯对照品(批号111524-201911,99.9%)和去氢木香内酯对照品(批号111525-201912,99.5%)均来源于中检院;15 批饮片分别购自四川(5 批)、云南(5 批)、广东(3 批)和广西(2 批)5 个省区,自制标准汤剂,批号分别为TJ01-TJ15。10 批样品由2 个生产厂家提供,批号见表3。
色谱纯乙腈和色谱纯磷酸;超纯水。
2 方法与结果
2.1 色谱条件
色谱柱信息:Scienhome YWG C18( P/N:96182540);Welch,XB-CN(Part Number:00205-31043);InertSustain AQ-C18(P/N:5020-89731);GL Sciences,Inertsil ODS-3(Cat.No.5020-01732);Agilent EXTEND C18(P.N.5188-5292),5 个厂家的色谱柱均为粒径5μm,长250mm,内径4.6mm。流动相:乙腈(A)-0.05%磷酸溶液(B)梯度洗脱(0~5min,32%~50%A;5~35min,50%~68%A;35~36min,68%~32%A,36~46min,32%A;在225nm 的波长处检测;30 ℃的柱温;体积流量:1.0mL/min,进样体积10μL;仪器显示的去氢木香内酯的理论塔板数应不低于3000。
2.2 标准汤剂的制备
取15 批次木香药材各200g,加水2000mL 煎煮1 小时,过滤,取滤渣加水1400mL 煎煮1 小时,过滤,合并滤液,低温蒸干,15 批次平均出膏86.3g,平均出膏率为43.2%。
2.3 对照品溶液
称取去氢木香内酯对照品、木香烃内酯对照品适量,精密称定,分别加甲醇溶解稀释成每1mL 含去氢木香内酯194.4μg、木香烃内酯213.5μg 的溶液,用孔径为0.45μm 的nylon 过滤膜过滤即得。
2.4 供试品溶液
分别精密称取木香对照药材0.25g;标准汤剂和配方颗粒,研细(四号筛)约0.5g;置150mL 带有瓶塞的锥形瓶中,用大肚吸管精密量取甲醇50mL,加入锥形瓶中,用电子天平称定重量后,超声波提取( 功率260 瓦,频率5 万赫兹)30min,放冷,用0.45μm 的nylon 过滤膜过滤,取续滤液,即得。
2.5 含量测定方法学考察
2.5.1 专属性
取“2.3~2.4”项下的溶液各10μL 注入液相色谱仪,结果去氢木香内酯峰与其它峰的分离度均大于1.5,且峰型好,表明专属性强,见图1。

图1 HPLC 色谱图(A)去氢木香内酯对照品(B)木香烃内酯和去氢木香内酯混合对照品(C)木香药材(D)木香配方颗粒(E)标准汤剂
2.5.2 线性关系考察
精密量取“2.3”项下去氢木香内酯对照品溶液,用移液枪分别精密量取4、10、50、80、200、400μL 置1mL 的量瓶中,分别加甲醇稀释至刻度,配制成系列浓度的标准溶液。分别进样10μL,以对照品的进样浓度(μg/mL)A 为横坐标,以去氢木香内酯峰面积积分值B 为纵坐标进行线性回归,建立标准曲线,去氢木香内酯的回归方程为A=15450 B-1151.4 (相关系数为0.9998) ,线性范围为0.656~65.6μg/mL。
2.5.3 精密度
取“2.4”项下的同一供试品溶液(批号:20090511),连续进样6 次,进行测定。去氢木香内酯的RSD 为0.8%,小于2.0%,表明仪器的精密度良好。
2.5.4 重复性
取同一批木香配方颗粒(批号:20090511),研细,照“2.4”项下的方法同时称取6 份供试品,用同一方法配制成供试品溶液,进样分析,计算去氢木香内酯的含有量RSD 为1.2%,小于2.0%,结果显示该提取方法和仪器方法的重复性良好。
2.5.5 准确度
分别称取已知含量的木香配方颗粒(批号:20090511)研细,混匀,取9 份,每份约0.25g,精密称定,每三份为一组,分别按样品中含量的50%,100%,150%的量加入去氢木香烃内酯对照品,按“2.4”项下的方法进行提取制备。按上述“2.1”项下描述的色谱条件测定并计算回收率。结果回收率 RSD 值为0.7%,小于3.0%,见表1。表明该提取方法可行。

表1 回收率实验结果(n=9)
2.5.6 稳定性
取同一批木香配方颗粒(批号:20090511)的供试品溶液,在室温(26℃)下放置,分别于0、1、2、3、6、12、18、24、48h 按上述“2.1”项下描述的色谱条件进样9 次,结果去氢木香内酯含量的RSD 为0.8%,低于3.0%,表明制备好的溶液在26℃下可以放置48h。
2.5.7 耐用性
分别采用岛津LC-30AT 型和Waters e2695-2998 型高效液相色谱仪,采用Scienhome YWG、Welch XB-CN、InertSustain AQ-C18、GL Sciences和Agilent EXTEND 5 个厂家的色谱柱按拟定的方法测定,结果去氢木香内酯含量的RSD 为1.8%,表明不同厂家的仪器和色谱柱检测效果良好。
2.5.8 样品的测定
取10 批木香配方颗粒,按前述“2.4”项下描述的方法制备供试品溶液,按前述“2.1”项下描述的色谱条件测定并计算样品中去氢木香内酯的含量,结果见表2。
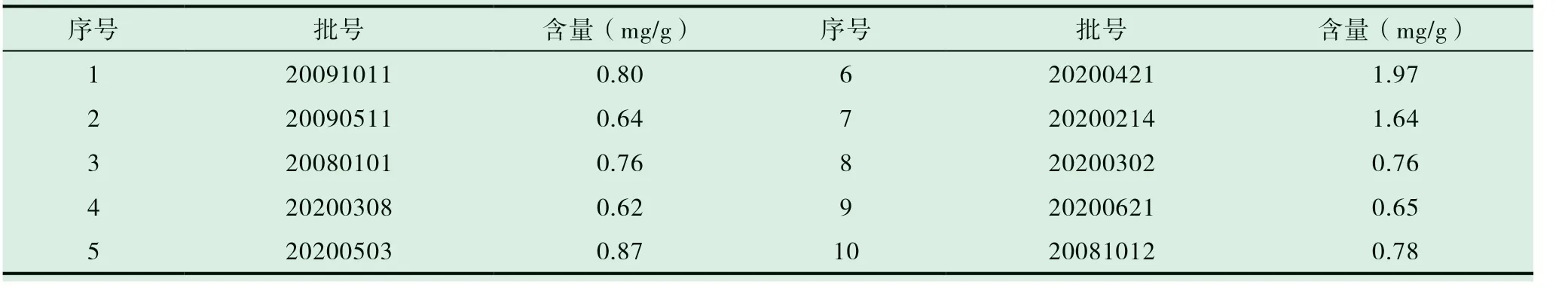
表2 样品测定结果(n=2)
2.6 指纹图谱方法学考察
2.6.1 精密度
取同一份批号为20090511 的木香配方颗粒供试品溶液,按前述“2.1”项下描述的色谱条件批处理进样6 次,进行测定,以3 号色谱峰(去氢木香内酯)为参照计算6 次色谱图中每个共同出现的色谱峰的相对保留时间、相对峰面积值的RSD,结果分别为0.7%,1.1%。表明7 个共有峰的相对保留时间RSD 与相对峰面积RSD 均小于2.0%,说明精密度良好。见图3。
2.6.2 重复性
取同一批木香配方颗粒(批号:20090511),混匀,0.5g 共6 份,精密称定,照“2.4”项下的提取方法配制供试品溶液,按前述“2.1”项下描述的色谱条件批处理进样分析,计算相对峰面积值和相对保留时间的RSD 值。结果7 个共有峰与参照峰的相对峰面积值和相对保留时间的RSD 均低于2.0%,表明方法重复性良好。
2.6.3 稳定性
取同一份木香配方颗粒供试品溶液(批号:20090511),在室温(26℃)下放置,分别于0、1、2、3、6、12、18、24、48h 按上述“2.1”项下描述的色谱条件进样9 次,以去氢木香内酯峰为参照峰,计算7 个共有峰的相对保留时间和相对峰面积的RSD,结果均小于2.0%,表明制备好的溶液在26℃下可以放置48h。
2.6.4 耐用性
分别采用LC-30AT 型和e2695-2998 型高效 液 相 色 谱 仪,采 用Scienhome YWG、Welch、InertSustain、GL Sciences、Agilent EXTEND 5 个牌子的色谱柱按拟定的方法测定,以去氢木香内酯峰为参照峰,计算7 个共有色谱峰的相对峰面积值和相对保留时间的RSD,结果均低于2.0%,结果显示方法的仪器及色谱柱耐用性良好。
2.7 指纹图谱的建立
2.7.1 分别精密吸取不同批次(S1-S10)的木香配方颗粒供试品溶液、混合对照品溶液、标准汤剂溶液和对照药材溶液批处理进样10μL,按“2.1”的色谱条件测定,记录色谱图,见图1。将10 批样品的实验数据导入国家药典委员会的中药色谱指纹图谱相似度评价系统(2012),采用中位数法对10 批样品的指纹图谱进行峰点校正,数据匹配分析,生成共有模式图及对照指纹图谱,见图2、3,确认7 个共有峰为特征峰,并用对照品图谱进行定位,确定2 号峰为木香烃内酯,3 号峰为去氢木香内酯,见图3。以3 号峰为参照峰,计算各共有峰的相对保留时间和相对峰面积,结果见表3、4。10 批木香配方颗粒与对照指纹图谱的相似度分别为0.989,0.989,0.982,0.990,0.989,0.989,0.972,0.972,0.973,0.980 相似度评价表明,10 批供试品指纹图谱和对照品指纹图谱的相关性良好,且7 个共有峰与标准汤剂中的峰都能一一对应。对比相似度较小样品与其他样品的指纹图谱,发现其共有峰相对保留时间比较一致,主要差异表现在色谱峰的面积上,即含量的差异,为不同厂家的样品。表明厂家不同样品的质量存在一定的差异。
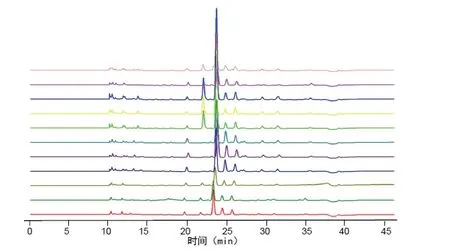
图2 10 批样品指纹叠加图谱
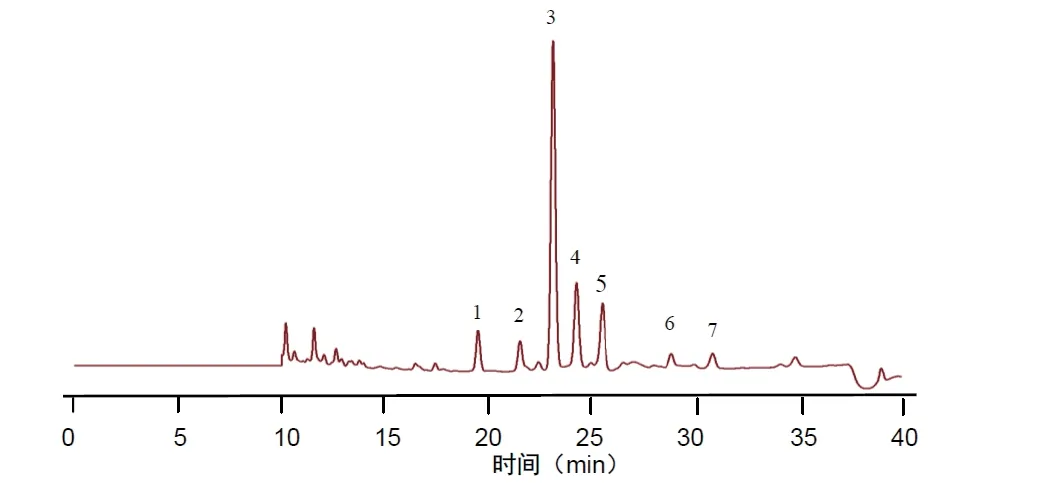
图3 对照指纹图谱
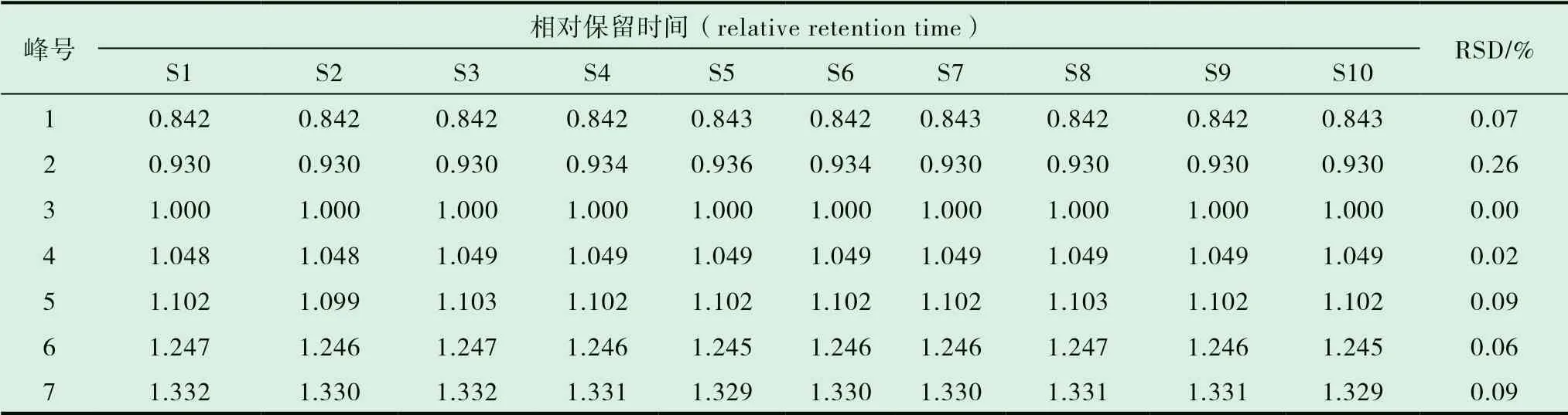
表3 10 批木香配方颗粒共有峰的相对保留时间
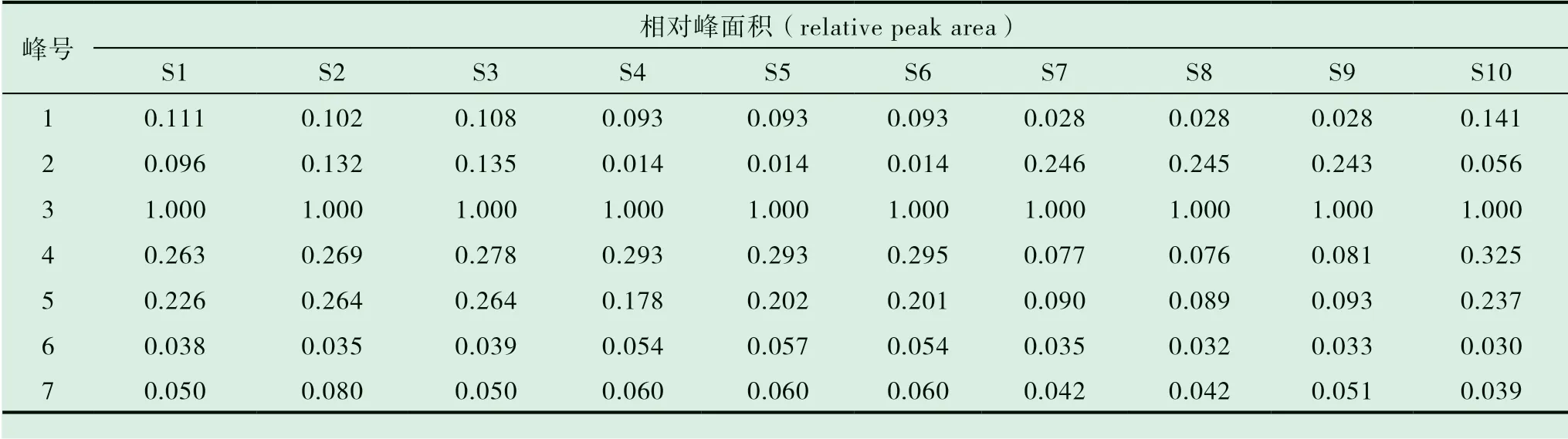
表4 10 批木香配方颗粒共有峰的相对峰面积
2.8 化学模式识别
2.8.1 聚类分析
以指纹图谱中标定的7 个共有峰的相对峰面积为变量,采用SPSS 19.0 软件中的平方Eudidean距离对10 批样品进行系统聚类分析,结果见图4。根据聚类分析结果,以类间距为指标,当聚类标定距离定为25 时,10 批样品聚为一大类,当聚类重新标定距离为4 时,样品S7、S8、S9 聚为第二小类,其余样品聚为第三小类。该分析与相似度评价结果一致。
2.8.2 主成分分析
对10 批样品进行主成分分析,以指纹图谱中标定的7 个共有峰峰面积为分析变量,将其导入SPSS 19.0 统计软件进行主成分分析,得到主成分特征值和方差贡献率,见表5,图5。可见前2 个主成分的特征值均大于1,累积贡献率为98.453%,基本可以反映各主成分的全部信息。
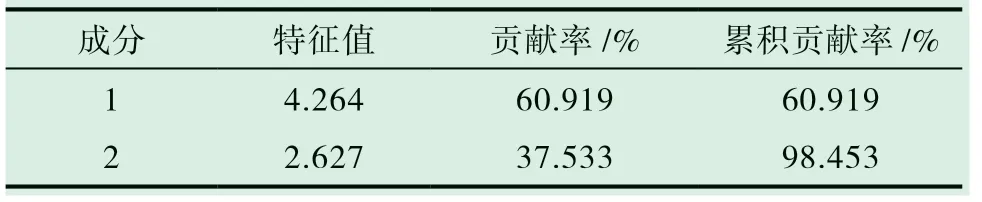
表5 主成分特征值和贡献率
2.8.3 综合评价
以各主成分对应贡献率为权重系数,计算10 批样品的主成分得分和综合得分,并进行排序,结果见表6。综合得分排名为S9>S5>S6>S4>S8>S7>S1>S2>S10>S3。其中生产日期较近的批次主成分得分较高,生产日期较远的批次相对较低,另外,不同样品批号中的药材产地不同也导致其质量有所不同。
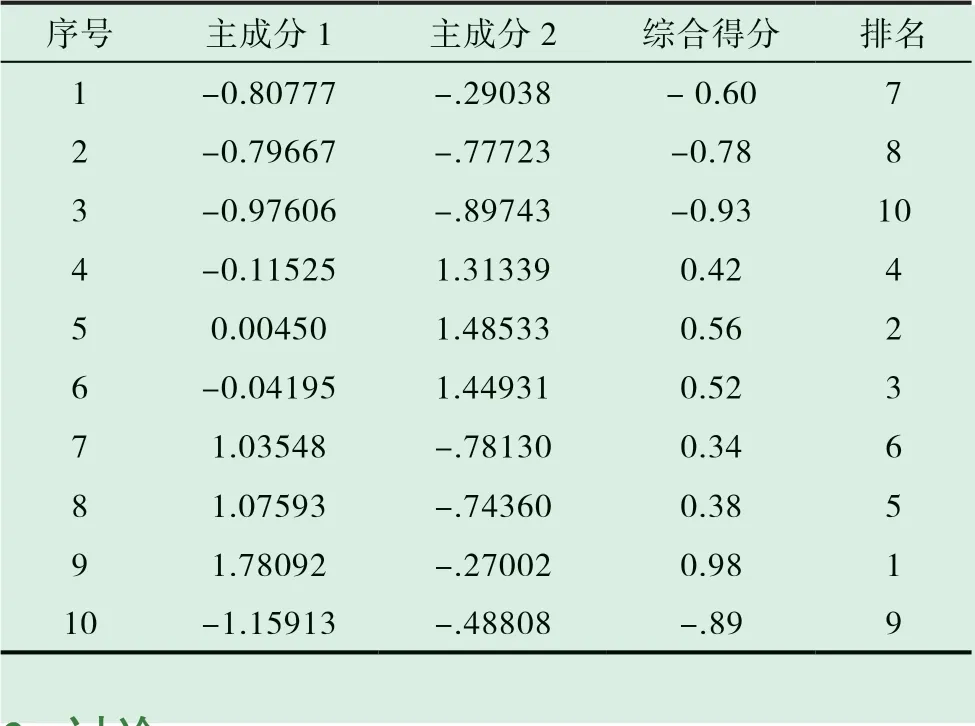
表6 10 批样品的主成分得分、综合得分和排名统计
3 讨论
实验过程中比较了甲醇、乙醇、70%甲醇、70%乙醇作为提取溶剂,结果甲醇提取效率最好;考察了甲醇-水、乙腈-水、乙腈-0.05%磷酸等流动相体系,考察了高效液相色谱仪和超高效液相色谱仪的检测效果,最终确定了文中的方法,该条件下色谱峰信息丰富,分离度好,稳定性高,可以作为控制木香配方颗粒的方法。
本实验通过高效液相色谱法对比研究木香药材,标准汤剂和市售配方颗粒,找到指标成分进行含量测定,并进行指纹图谱的研究及相似度评价,对木香配方颗粒进行了定性、定量评价,相似度都大于0.97,表明其质量比较一致,且与标准汤剂图谱也一致,可见该方法更具可靠性。但是通过与药材图谱进行对比后发现制成配方颗粒后木香烃内酯的含量下降较多,故没有选取木香烃内酯进行含量测定。而且其他峰也有变化,表明药材与配方颗粒在成分方面有一定的差异,还有待研究。
采用聚类分析将10 批样品分为3 类,主成分分析采用降维模式,将反映HPLC 指纹图谱的多维特征参数用2 个主成分表示,可为木香配方颗粒质量评价提供参考。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
CHINA TODAY(2022年8期)2022-08-03
小小说月刊(2022年14期)2022-07-18
中国农业科学(2022年10期)2022-06-28
今日农业(2021年2期)2021-11-27
科学导报(2021年33期)2021-06-07
作文大王·低年级(2020年2期)2020-03-13
思维与智慧·下半月(2017年9期)2017-09-29
食品工业科技(2014年13期)2014-03-11
中国医药导报(2011年27期)2011-12-31
