
一测多评法测定黑果腺肋花楸果中三种花色苷的含量①
2022-06-07 07:42张婷婷李兰兰李新霞
新疆医科大学学报 2022年5期
张婷婷,李兰兰,李新霞
(新疆医科大学药学院,乌鲁木齐830011)
黑果腺肋花楸果(Black chokeberry)又称不老莓,是蔷薇科腺肋花楸属植物黑果腺肋花楸[Aronia melanocarpa(Michx.)Elliott]的果实,2018年9月国家卫健委正式批准黑果腺肋花楸果为新型食品原料[1]。黑果腺肋花楸果实中主要化学成分为花青素、黄酮、多酚[2-3]等,花青素是黑果腺肋花楸果中最主要的多酚类物质,在植物体内多以糖苷形式存在,称为花色苷,主要由矢车菊素-3-O-半乳糖苷(Cyanidin-3-OGalactoside,C3Ga)、矢车菊素-3-O-阿拉伯糖苷(Cyanidin 3-O-arabinoside,C3A)、矢车菊素-3-O-葡萄糖苷(Cyanidin-3-O-glucoside,C3G)和矢车菊素-3-O-木糖苷组成[4]。近年来大量的研究表明黑果腺肋花楸果花色苷提取物具有较强的抗氧化[5-6]、抗衰老[7]、抗癌[8]、降血糖血脂[9-10]等活性。目前多采用pH 示差法[11-12]、高效液相色谱外标法[13-14]测定花色苷含量。pH示差法虽然价格便宜但只适用于检测总花色苷含量,且易受辅色素转化等因素影响,引起误差;高效液相色谱外标法同时对多个花色苷成分进行测定时价格昂贵成本较高。一测多评法(Quantitative analysis of multi-components by single-marker, QAMS)是可用于多成分质量控制,通过只测定某个(易得、价格较低、有效)代表性成分,同时计算出其它待测有效成分的含量的方法[15-17],该方法操作简单,成本较低。本研究在建立外标法的基础上,采用一测多评法以价格相对较低的矢车菊素-3-O-葡萄糖苷为内参,建立另外两种成分的校正因子并计算含量,为黑果腺肋花楸果质量评价提供一种低成本的方法。
1 仪器与试药
1.1 仪器LC-2030C 高效液相色谱仪及色谱工作站(日本岛津公司);Agilent 1260高效液相色谱仪及色谱工作站(美国安捷伦科技公司);150 W,40 kHZ 超声波清洗仪(昆山市超声仪器有限公司);AB135-S 分析天平(梅特勒-托利多仪器上海有限公司);Exceed-EUV 纯水仪(成都唐氏康宁科技发展有限公司);SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司);AQ-C18(250 mm×4.6 mm, 5 µm)、Gensial-C18(250 mm×4.6 mm, 5 µm)、Agilent-C18(250 mm×4.6 mm,5 µm)。
1.2 试药C3Ga(北京索莱宝科技有限公司,批号:220G021)、C3G(北京索莱宝科技有限公司,批号:1217N021)、C3A(北京索莱宝科技有限公司,批号:311F021)、色谱乙腈(美国Merck)、盐酸、甲醇、甲酸为分析纯。
1.3 药材黑果腺肋花楸果粉由新疆埃乐欣药业有限公 司 提 供(批 号:S1:20210103、S2:20210118、S3:20210223、S4:20210318、S5:20210425、S6:20210508)。
2 方法与结果
2.1 溶液的制备
2.1.1 对照品溶液的制备 精密称取C3Ga、C3G、C3A对照品各适量,用甲醇(含2%HCl,V/V)配制成浓度分别为90.00、9.80、60.00µg/mL的花色苷混合对照品母液,经稀释配制成C3Ga、C3G、C3A 系列浓度分别为7.00、11.66、19.44、32.40、54.00、90.00 µg/mL;0.76、1.27、2.12、3.53、5.88、9.80 µg/mL;4.67、7.78、12.96、21.60、36.00、60.00µg/mL的混合对照品溶液。
2.1.2 供试品溶液的制备 称取黑果腺肋花楸果粉1.0 g,用80%含酸甲醇(0.8% HCL 调节pH 至2~3),料液比1∶10,40 ℃条件下超声提取40 min,冷却至室温后滤过,取滤液1 mL定容至10 mL即得。
2.2 色谱条件色谱柱:AQ-C18柱(250 mm×4.6 mm,5µm);流动相:乙腈(A)-0.5%甲酸(B),梯度洗脱(0~15 min, 10%A~14%A; 15~20 min, 14%A~16%A; 20~30 min, 16%A~19%A; 30~35 min, 19%A; 35~40 min,19%A~10%A);流速:1.0 mL/min;检测波长:520 nm;柱温:30 ℃;进样量:20µL,见图1。

图1 混合对照品(A)和黑果腺肋花楸果供试品(B)色谱图
2.3 外标法方法学考察
2.3.1 回归方程的建立 分别吸取“2.1.1”项下C3Ga、C3G、C3A 系列浓度混合对照品溶液,按“2.2”项下色谱条件进样,以浓度为X轴,峰面积为Y轴,得到C3Ga、C3G、C3A 的线性回归方程,结果C3Ga 的回归方 程 为Y= 59454X- 3212.5,r=0.9998,浓 度 在7.00~90.00µg/mL范围内线性关系良好;C3G的回归方 程 为Y= 73673X- 164.77,r=0.9998,浓 度 在0.76~9.80 µg/mL 范围内线性关系良好;C3A 的回归方程为Y= 76238X- 3621.2,r=0.9998,浓度在4.67~60.00µg/mL范围内线性关系良好。
2.3.2 日内间精密度 分别精密吸取C3Ga、C3G、C3A的低、中、高浓度混合对照品溶液,按“2.2”项下色谱条件进样,重复进样3 次,连续3 d,记录C3Ga、C3G、C3A的峰面积,计算日内间精密度,见表1。
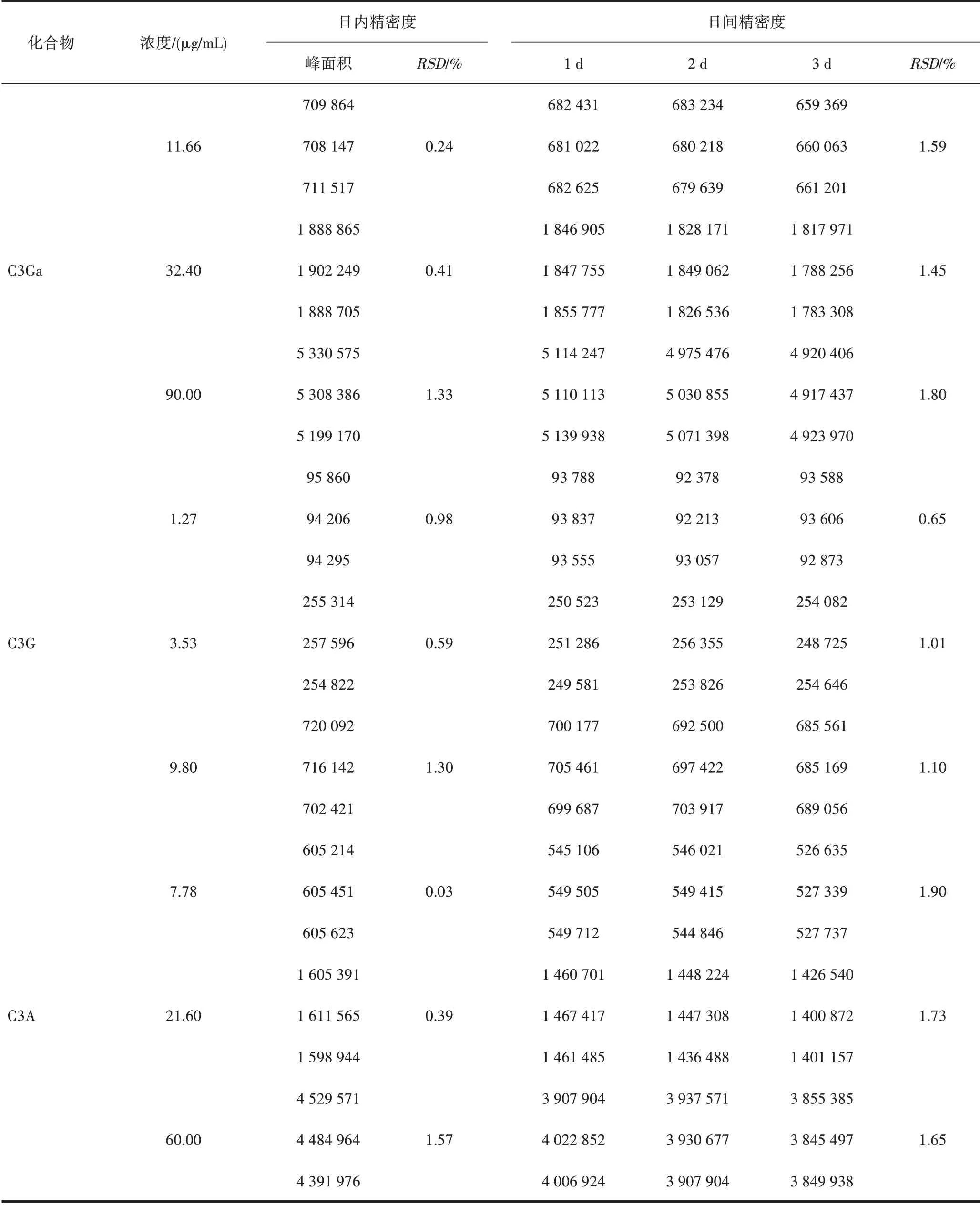
表1 日内间精密度
2.3.3 重复性试验 按“2.1.2”项下操作,提取同一批黑果腺肋花楸果粉供试品溶液(n=6),按“2.2”项下色谱条件测定,记录C3Ga、C3G、C3A 色谱峰面积,计算得到C3Ga、C3G、C3A 峰面积的RSD分别为0.60%、1.26%、0.96%,表明方法重复性良好。
2.3.4 稳定性试验 吸取“2.1.2”项下供试品溶液于0、2、4、6、8、12和24 h依法进样,按“2.2”项下方法测定,分别记录C3Ga、C3G、C3A 峰面积并计算RSD,RSD分别为0.70%、0.92%、0.87%,表明C3Ga、C3G、C3A 溶液在24 h内稳定。
2.3.5 回收率试验 分别吸取供试品溶液适量,以0.8∶1、1:1、1.2:1 加入C3Ga、C3G、C3A 对照品溶液,依法测定,经计算C3Ga、C3G、C3A 对照品的加样回收率分别为1.92%、1.70%、1.01%,见表2。
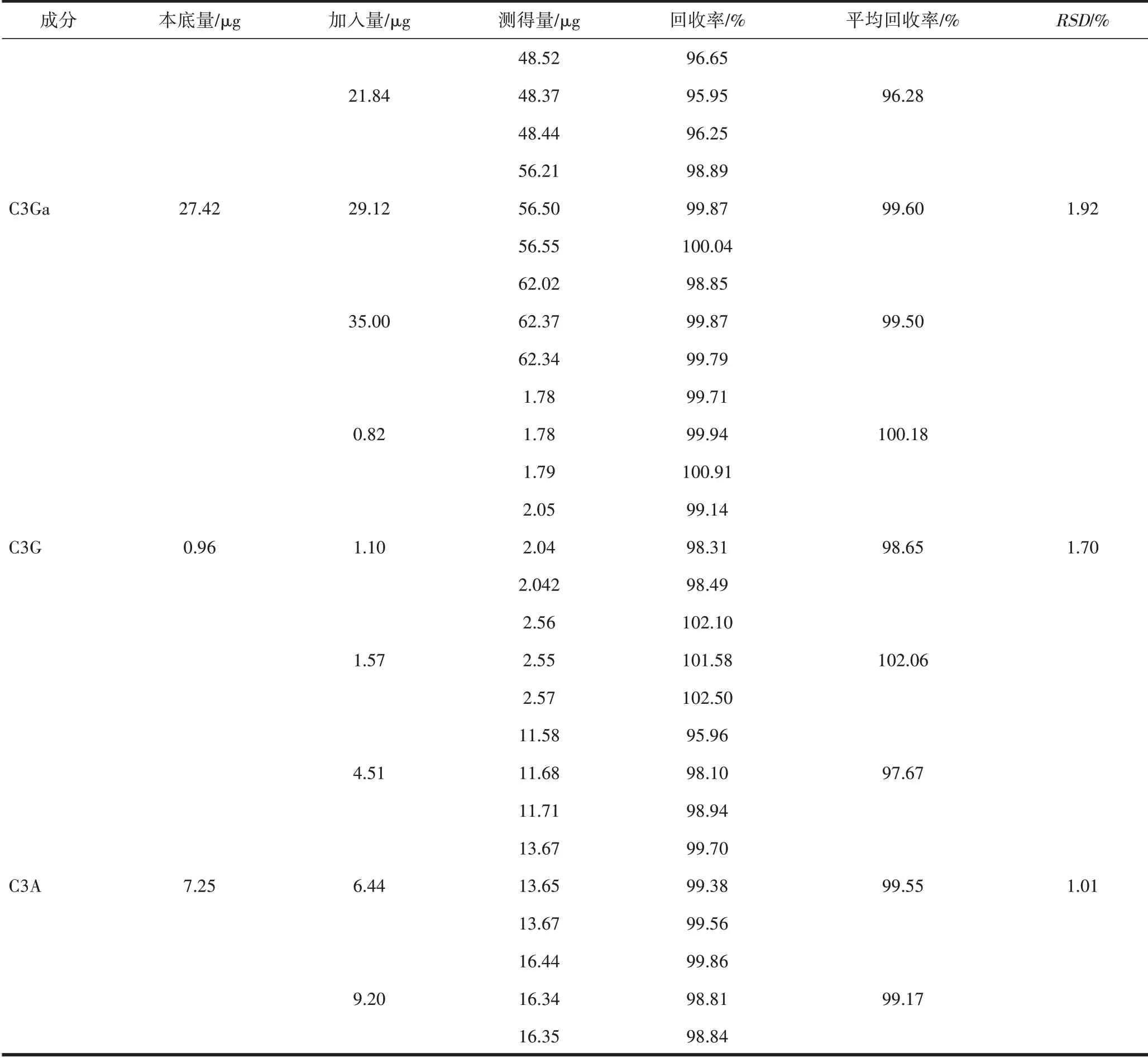
表2 回收率试验
2.4 相对校正因子的建立精密吸取“2.1.1”项下系列浓度混合对照品溶液各适量,按“2.2”项下方法测定,以C3G 为内参,按公式:f=AS×Ci/Ai×Cs式中:Ci 为C3G 浓度,Ai 为C3G 峰面积,Cs 为待测组分浓度,As为待测组分峰面积计算C3Ga、C3A 的相对校正因子,见表3。

表3 相对校正因子结果
2.5 校正因子耐用性考察
2.5.1 不同检测波长的影响 精密吸取“2.1.1”项下浓度分别为54.00、5.88、36.00 µg/mL 的C3Ga、C3G 和C3A 混合对照品溶液(n=3),按“2.2”项下方法测定,记录不同波长(518、520、522 nm)下的峰面积,以C3G 色谱峰为内参,计算相对校正因子。C3Ga、C3A 相对校正因子均值分别为1.2671、1.0465,RSD分别为0.024%、0.16%。表明当检测波长在518~522 nm范围内波动时,校正因子不受影响。
2.5.2 不同柱温对校正因子的影响 精密吸取“2.1.1”项下浓度分别为54.00、5.88、36.00 µg/mL 的C3Ga、C3G 和C3A 混合对照品溶液(n=3),按“2.2”项下方法测定,记录不同柱温(25、30、35 ℃)下的峰面积,以C3G 色谱峰为内参,计算相对校正因子。C3Ga、C3A相对校正因子均值分别为1.2545、1.0664,RSD分别为1.01%、1.85%。表明当柱温在25~35 ℃范围内波动时,对校正因子的测定影响不大。
2.5.3 不同流速对校正因子的影响 精密吸取“2.1.1”项下浓度分别为54.00、5.88、36.00 µg/mL 的C3Ga、C3G 和C3A 混合对照品溶液(n=3),按“2.2”项下方法测定,记录不同流速(0.8、1.0、1.2 mL/min)下的峰面积,以C3G 色谱峰为内参,计算相对校正因子。C3Ga、C3A相对校正因子均值分别为1.2443、1.0687,RSD分别为1.58%、1.86%。表明流速在0.8~1.2 mL/min对校正因子无显著影响。
2.5.4 仪器与色谱柱的考察 精密吸取“2.1.1”项下浓度分别为54.00、5.88、36.00 µg/mL 的C3Ga、C3G 和C3A 混合对照品溶液(n=3),按“2.2”项下方法测定,分别采用岛津LC-2030C,Agilent 1260 高效液相色谱仪,Agilent XDB-C18、AQ-C18、Gensial-C18色谱柱依法测定,记录C3Ga、C3G、C3A 峰面积,以C3G 色谱峰为内参,计算相对校正因子。C3Ga、C3A 相对校正因子分别为1.2576、1.0714,RSD分别为1.02%、2.72%。表明相对校正因子受仪器与色谱柱影响不显著,见表4。

表4 不同仪器与色谱柱对校正因子的影响
2.6 待测色谱峰的定位精密吸取“2.1.1”项下浓度分别为54.00、5.88、36 .00 µg/mL 的5 号C3Ga、C3G 和C3A 混合对照品溶液(n=3),按“2.2”项下方法测定,考察不同仪器与色谱柱对C3Ga、C3A 相对保留时间的影响。结果表明各成分相对保留时间受仪器与色谱柱影响较小,见表5。

表5 不同仪器与色谱柱对保留时间的影响
2.7 样品含量测定取不同批次黑果腺肋花楸果粉,按“2.1.2”项下制备供试品溶液(n=3),按“2.2”项下方法测定,记录各批次供试品中C3Ga、C3G、C3A 色谱峰面积,分别采用外标法和QAMS法计算不同批次黑果腺肋花楸果粉中花色苷含量,外标法测定6批黑果腺肋花楸果中C3G 含量分别为0.139、0.148、0.131、0.204、0.088、0.159 mg/g;C3Ga 分别为3.899、4.184、3.698、5.855、2.334、4.537 mg/g;C3A 分 别 为1.070、1.142、1.009、1.597、0.641、1.239 mg/g;QAMS 法测定6批黑果腺肋花楸果中C3Ga 分别为3.898、4.196、3.714、5.879、2.344、4.561 mg/g;C3A 分 别 为1.068、1.143、1.012、1.602、0.641、1.244 mg/g。Pearson相关性分析表明两种方法所测数据相关系数为1.000,P值为0.000,测定结果具有显著相关性,说明建立的一测多评方法结果准确,见表6。

表6 Pearson相关性分析结果
3 讨论
前期实验对花色苷类成分进行前处理考察,发现花色苷类成分在酸性甲醇中提取效率更高,因此后续以80%甲醇(0.8%HCl,V/V),超声40 min,料液比1∶10,提取1次为最佳前处理条件。本实验采用梯度洗脱建立黑果腺肋花楸果3种花色苷类成分HPLC法,本方法与文献[14]相比化合物分离度较好,3种成分方法学验证均符合要求。在此基础上以最为常见且价格较低的矢车菊素-3-O-葡萄糖苷对照品为内参,建立一测多评法测定黑果腺肋花楸果中3 种花色苷类成分含量。经典外标法虽可对3 种花色苷进行测定,但由于使用多种花色苷类对照品成本较高,而一测多评法可用一种花色苷对照品利用相对校正因子对多种花色苷成分进行定量,并根据相对保留时间进行定性,实现以一种成分同时对多种成分进行定性定量的目的。花色苷类成分最大吸收波长在500~550 nm 之间,在此波长范围内干扰较小,在520 nm 最大吸收波长进行检测时,矢车菊素-3-O-半乳糖苷、矢车菊素-3-O-葡萄糖苷、矢车菊素-3-O-阿拉伯糖苷成分响应更高,峰面积更大,可用于定量分析。另外还发现另一花色苷类成分,经文献查阅后,认为可能是矢车菊素-3-O-木糖苷,后续可采用液质联用进一步指认。
一测多评法由王智民等[18]于2006年首次提出,目前多用于解决对照品稀有、昂贵,缺乏对照品或多指标评价中药质量等方案,2020 版《中国药典》采用一测多评法收载品种有黄连、穿心莲、银杏叶片[19]等药材及制剂,该方法也被美国药典等所采纳。黑果腺肋花楸果中富含多种花色苷,但花色苷类成分不稳定且价格较高,不易获得,采用一测多评法可实现多种花色苷类成分的同步测定,低成本为黑果腺肋花楸果中花色苷质量评价提供新方法。
猜你喜欢
人民长江(2021年9期)2021-10-18
少儿画王(3-6岁)(2021年8期)2021-09-12
学苑创造·A版(2021年4期)2021-04-18
小学阅读指南·低年级版(2021年3期)2021-03-19
课堂内外(高中版)(2021年7期)2021-01-17
初中生学习指导·提升版(2020年10期)2020-09-10
大众摄影(2018年6期)2018-06-19
红蜻蜓(2017年8期)2017-10-30
佛山陶瓷(2017年8期)2017-09-06
儿童时代·快乐苗苗(2016年5期)2016-09-14
