
ExoCET-BAC策略高效抓取和组装高AT含量基因组大片段
2022-03-16 03:24姜婵娟崔天琦孙洪娈焦念志符军张友明王海龙
合成生物学 2022年1期
姜婵娟,崔天琦,孙洪娈,焦念志,符军,张友明,王海龙
(1 山东大学微生物技术研究院,微生物技术国家重点实验室,山东 青岛266237; 2 山东大学海洋研究院,山东 青岛266237)

随着测序技术的不断进步,越来越多的微生物基因组被测序,其中蕴含的众多未知生物合成资源亟待开发。为此人们陆续开发了靶向抓取基因组大片段的方法,包括:基于酵母内源同源重组的TAR 技术[1-2],基于大肠杆菌噬菌体同源重组酶的RecET 技术[3],基于CRISPR/Cas9 体 外酶切和Gibson 体外组装的CATCH 技术[4-5],基于核酸外切酶介导的体外同源重组和大肠杆菌RecET 蛋白介导的细胞内同源重组联用的ExoCET 技术[6],基于CRISPR/Cas9 体外酶切和体外λ 包装相结合的方法[7],基于Cas12a 体外酶切和大肠杆菌细胞内Cre-lox 位点特异性重组的CAPTURE 技术[8]。除了基因组大片段直接抓取外,人们还开发了一些将大型基因簇拆分为若干DNA 片段,然后先利用PCR 扩增这些片段,再将它们拼接起来形成完整基因簇的克隆技术,比如DiPaC[9-10]、CPEC[11]、DNA Assembler[12-13]等。然而,近年来人们大多针对次级代谢产物丰富的放线菌等高GC 含量微生物对这些技术进行条件优化,却忽略了对高AT 含量大型基因簇克隆技术的开发,导致高AT 含量基因簇克隆困难。
在DNA 双螺旋结构中,配对碱基之间的氢键通过持续断裂和再生的动态平衡而发生DNA 呼吸作用[14]。由于AT 之间的氢键相互作用与GC 相比较弱,因此高AT 含量DNA 片段的呼吸作用较强,导致其中含有较多的单链泡状结构,基因表达相关的蛋白更容易结合在这些区域,使其中基因的转录活性较强。这是导致高AT含量DNA较不稳定和克隆困难的重要原因。为此,Godiska 等[15]建立了利用线性pJAZZ 载体在大肠杆菌中克隆高AT含量DNA 的方法,但是此载体的最大容量仅为30 kb。前述CATCH 等克隆技术虽然能从基因组上抓取大于30 kb 的基因组大片段,但是它们没有针对高AT 含量DNA 进行优化,因此在克隆高AT 含量基因簇时遇到了困难,比如CATCH 技术无法从AT含量为63%的变形链球菌(Streptococcus mutans)基因组中克隆DNA 大片段[16]。中国科学院微生物研究所陈义华教授课题组[16]针对高AT 含量厌氧菌来源次级代谢产物生物合成基因簇克隆的瓶颈,利用变形链球菌UA159 菌株能够天然转化外源DNA 和具有较高同源重组活性的特点开发的NabLC 大片段克隆技术,能将40 kb 以内的高AT含量厌氧菌来源的基因簇整合到UA159 基因组的特定位置上,进而成功克隆了变形链球菌(AT 含量63%)、表皮葡萄球菌(AT 含量68%)和梭菌(AT 含量72%)的基因簇,并在UA159 菌株中进行了这些基因簇的异源表达研究。但是利用NabLC 技术克隆的基因簇整合在变形链球菌UA159 菌株的基因组上,因此,只能在UA159 菌株中进行表达研究。如果基因簇在UA159 菌株中不表达,则无法将其转移到其他宿主中进行表达优化。
ExoCET 技术是通过联合核酸外切酶(exonuclease)介导的体外同源重组和大肠杆菌RecET蛋白介导的细胞内同源重组,将基因组大片段从限制性酶切或CRISPR/Cas9 酶切的基因组中一步抓取到载体上的克隆技术[6]。ExoCET 是在RecET[3]的基础上发展起来的基因组大片段克隆技术。噬菌体蛋白RecE 是5'→3'双链DNA 核酸外切酶,RecT 是单链DNA 退火蛋白。双链线状DNA 在RecE 的作用下暴露出3'单链末端,然后RecT 介导单链末端通过碱基互补配对进行退火,使DNA 片段首尾相接串联起来[3]。基于上述原理开发的RecET 技术可以从基因组上将50 kb 以内的目的DNA 片段抓取到表达载体上[3]。RecET 技术要求克隆载体和目的DNA 片段必须同时进入一个宿主细胞内并相遇才能发生同源重组。受到当前基因组制备方法的限制,基因组DNA 在制备过程中容易断裂的特点致使越大的目的DNA 片段在制备的基因组中含量越低,转化时其与克隆载体进入同一细胞的概率也就越小,因此克隆效率也就越低。所以RecET 技术不能从细菌基因组中克隆大于50 kb 的DNA 片段[3]。ExoCET 技术靶向抓取基因组大片段分为两个过程,首先体外同源重组将载体和目的DNA 片段串联起来提高其一起进入克隆宿主的概率,然后细胞内表达的RecET 蛋白将只完成了一端退火的重组DNA 分子(占体外反应产物的85%)通过另一端的同源臂进行退火,形成环状重组质粒[6]。这种体外同源重组和细胞内同源重组相结合的方法,使基因组大片段的克隆效率比RecET技术高80多倍。ExoCET能以超过80%的正确率将>50 kb的非核糖体多肽合成酶基因从发光杆菌基因组克隆到质粒载体上,也能将106 kb的盐霉素聚酮合酶基因簇以4%~8%的正确率从限制性酶切或CRISPR/Cas9 酶切的白色链霉菌基因组克隆到BAC 载体上[6]。而且,利用该ExoCET技术克隆的基因簇还能在大肠杆菌中通过丰富的Red/ET 重组[17-18]工具箱进行修饰,以插入基因转移元件[19-20]、替换启动子[21-22]、删除/插入调控基因[23]等实现在多种不同宿主的异源表达,而且还可以进行结构域点突变或模块替换进行组合生物合成以获得更多衍生物[24-26]。目前尚未有利用ExoCET 技术从AT 含量>63%的基因组中抓取大片段的报道。由于ExoCET 技术通过联合体外同源重组和细胞内同源重组,能够最大限度地促使克隆载体和目的基因组片段同时进入一个宿主细胞并发生同源重组,因此具有很高的克隆效率。如果对ExoCET 技术进行优化,使其能够高效克隆高AT含量基因,将非常有利于高AT含量生物的基因组功能研究。
本研究以AT 含量为69%的海洋单细胞光合蓝细菌原绿球藻(Prochlorococcus)MIT 9301菌株的基因组为研究对象,探索了利用ExoCET 技术进行高AT含量基因组大片段克隆的可行性,并对相关操作条件进行了优化,成功建立了基于ExoCET 和单拷贝BAC 载体的ExoCET-BAC 策略,实现了大于80 kb基因组大片段的靶向抓取、11个片段的高效组装和4个基因组大片段的一步靶向抓取,为高AT含量生物的基因组功能研究提供了高效克隆技术。
1 材料和方法
1.1 材料
1.1.1 菌株、质粒和引物
本研究所用的菌株和质粒见表1。寡核苷酸引物(由睿博兴科生物技术有限公司合成)见表2。

表1 本研究所用的菌株和质粒Tab.1 Strains and plasmids used in this study

表2 本研究所用寡核苷酸序列Tab.2 Oligonucleotide sequences used in this study

续表

续表

续表
1.1.2 培养基
LB 培养基(1 L):胰蛋白胨10 g,酵母提取物5 g,氯化钠1 g。
Pro99培养基(1 L):贫营养海水1 L(0.22 μm混合纤维素微孔滤膜抽滤,121 ℃,20 min 高压灭菌),NaH2PO4·H2O[6.90 g/L,0.22 μm Millipore Express(PES)膜过滤器过滤除菌]2 mL,NH4Cl[42.80 g/L,0.22 μm Millipore Express(PES)膜过滤器过滤除菌]1.6 mL,微量元素溶液100 μL。
微量元素溶液:Na2EDTA·2H2O 1.17×10−6mol/L,FeCl3·6H2O 1.17×10−6mol/L,ZnSO4·7H2O 8.00×10−9mol/L,CoSO4·7H2O 5.00×10−9mol/L,MnCl2·4H2O 9.00×10−8mol/L,Na2MoO4·2H2O 3.00×10−9mol/L, Na2SeO31.00×10−8mol/L,NiCl2·6H2O 1.00×10−8mol/L,0.22 μm Millipore Express(PES)膜过滤器过滤除菌。
1.1.3 主要试剂和仪器
试剂:PrimeSTAR Max DNA Polymerase、Tks GflexTMDNA Polymerase 和DNA Markers 购 自Takara 公司;蛋白酶K 购自Roche 公司;蛋白胨和酵母粉购自OXOID 公司;琼脂粉、琼脂糖、25×TAE 和10×TBE 购自Solarbio 公司;RNase A 购自Thermo Scientific 公司;限制性内切酶、Gibson 组装克隆试剂盒和T4 DNA 聚合酶购自New England Biolabs 公司;DNA 纯化回收试剂盒和溶菌酶购自天根生化科技有限公司;DNA 提取液(苯酚∶氯仿∶异戊醇=25∶24∶1)购自北京鼎国昌盛生物技术有限责任公司。
仪器:杭州柏恒科技有限公司PCR仪,杭州奥盛仪器有限公司离心机和恒温混匀仪,Eppendorf电转化仪,美国Quawell超微量分光光度计。
1.2 方法
1.2.1 原绿球藻MIT 9301基因组制备
将50 mL培养7 d的MIT 9301培养液8000 r/min离心10 min,弃上清。用1 mL灭菌的ddH2O重悬菌体,吹打混匀并转入2 mL EP管中,10 000 r/min离心5 min,弃上清。加入400 μL SET 溶液(75 mmol/L NaCl,25 mmol/L EDTA,20 mmol/L Tris,pH 8.0)吹打混匀菌体。加入30 μL溶菌酶(20 mg/mL),颠倒混匀,37 ℃水浴2 h。加入30 μL 蛋白酶K(20 mg/mL),颠倒混匀后加入40 μL 10%SDS,颠倒混匀,50 ℃水浴1 h,直至溶液澄清。加入100 μL 5 mol/L NaCl,颠倒混匀。加入600 μL DNA提取液(苯酚∶氯仿∶异戊醇=25∶24∶1),颠倒混匀至溶液呈乳白色,10 000 r/min离心30 min,用剪掉枪尖的200 μL枪头取500 μL上清至新的2 mL EP管中。加入35 μL 3 mol/L 醋酸钠(pH 7.5)颠倒混匀,然后加入1.2 mL的无水乙醇颠倒混匀后,管中会出现絮状DNA。用200 μL枪头挑取絮状DNA并转入装有1 mL 75%乙醇的1.5 mL EP管中,10 000 r/min离心2 min,弃上清,倒置于吸水纸上,室温晾干至基因组呈透明状。加入350 μL ddH2O 溶解基因组DNA,测定浓度后4 ℃保存备用。
MIT 9301 基因组酶切:400 μL 酶切体系含有10 μg 基因组DNA,40 μL 10 × NEB Cutsmart 缓冲液,10 UPmeⅠ,2 μL RNaseA(10 mg/mL),37 ℃酶切4 h。酶切结束后用DNA 提取液(苯酚∶氯仿∶异戊醇=25∶24∶1)对酶切产物进行抽提,然后进行乙醇沉淀(具体过程同基因组提取步骤)。
1.2.2 PCR制备克隆载体和组装用DNA片段
PCR 扩增pBR322 克隆载体,以pBR322-ampccdB-rpsL[19]为模板,PCR产物长度为2114 bp。PCR扩增原绿球藻基因组片段,PCR长度为3000 bp。扩增pBeloBAC11克隆载体,以pBeloBAC11-hygccdB为模板,PCR产物长度为7501 bp。PCR反应所用的酶为PrimeSTAR Max DNA Polymerase。PCR 所用引 物 见 表2。PCR 反 应 体 系(50 μL):25 μL PrimeSTAR Max Premix(2×);Primer 1、Primer 2各2 μL;模板200 ng;ddH2O补齐至50 μL。PCR反应条件:94 ℃2 min;98 ℃10 s,Tm5 s,72 ℃5 s/kb,30 cycles;72 ℃5 min;4 ℃hold。
1.2.3 ExoCET直接克隆和多片段组装
①T4 聚 合 酶(T4pol) 反 应 体 系 载 体200 ng,PCR 扩增基因组片段200 ng 或PmeⅠ酶切 的MIT 9301 基 因 组DNA 10 μg,2 μL 10 ×NEB Buffer 2.1 和0.13 μL T4 DNA 聚 合 酶,用ddH2O 补齐至20 μL。混匀后放入PCR 仪中进行体外反应,具体程序为:25 ℃1 h,75 ℃20 min,50 ℃1 h,4 ℃hold。反应产物在室温下MF-Millipore 0.025 μm MCE 混合纤维素酯亲水膜脱盐40 min 备用。
②Gibson组装反应体系 DNA用量与T4聚合酶反应相同,DNA混合液与Gibson组装预混液等比例混匀后放入PCR仪中进行体外反应,具体程序为:50 ℃1 h,4 ℃hold。反应产物在室温下脱盐40 min备用。
③电转化感受态细胞制备 转接40 μL 过夜培养的含有pSC101-BAD-ETgA-tet 质粒的E.coliGB05-dir菌株培养液至1.3 mL 含有4 μg/mL 四环素的LB培养基中,30 ℃,950 r/min培养2 h,之后加入35 μL 10%L-阿拉伯糖溶液,37 ℃,950 r/min培养40 min。9500 r/min,离心1 min,收集菌体,加入1 mL ddH2O重悬后,9500 r/min,离心1 min,收集菌体,然后再重复洗一遍。之后将脱盐的T4聚合酶反应或Gibson 组装反应产物与菌体混匀后转入1 mm电转杯中,1350 V电击。然后将1 mL LB培养基加入电转杯中重悬菌体,再将菌液转入1.5 mL EP管中,复苏1 h后,9500 r/min,离心1 min收集菌体,涂布到含有相应抗生素的LB 平板上,37 ℃培养过夜。
2 结果和分析
2.1 ExoCET 组装高AT 含量DNA 片段时,体外重组使用Gibson较T4聚合酶能获得更高效率
首先为了测试ExoCET 组装高AT 含量DNA 的可行性,对PCR 扩增的4 个3 kb MIT 9301 基因组片段(4 个片段的AT 含量分别为69%、69%、73%、70%)和1 个2 kb pBR322 载体,共计5 个DNA片段,先在体外分别利用T4pol体系和Gibson体系进行处理,然后转化到表达RecET 重组酶的大肠杆菌中进行ExoCET 组装[图1(a)]。这5 个DNA 片段相互之间在末端带有40 bp同源臂。结果显示体外同源重组不论使用T4pol 体系还是Gibson体系均获得了87.5%的正确率[图1(b)],而且Gibson+RecET 的ExoCET 获得了更多的菌落数,是T4pol+RecET 的22 倍[图1(c)]。这 说 明ExoCET 能高效组装高AT 含量DNA 片段。在体外同源重组中,Gibson 反应体系中包含T5 核酸外切酶、Phusion DNA 聚合酶和Taq 连接酶,在这些酶的作用下形成的DNA 拼接产物是不含缺口的共价键连接的DNA 分子,而T4pol 反应获得DNA 拼接产物是带有缺口的DNA 分子。高AT 含量的DNA片段在体外同源重组中通过同源臂区碱基互补配对形成的共价键连接的双链DNA 分子与带缺口的双链DNA 分子与相比更稳定,因此体外重组使用Gibson较T4聚合酶能获得更高效率。

图1 ExoCET技术组装5个DNA片段(a)5个片段组装示意图;(b)组装获得重组质粒的EcoRⅠ酶切图谱,胶图右侧为酶切理论图谱,1~24为Gibson+RecET;25~48为T4pol+RecET;(c)Gibson+RecET和T4pol+RecET五片段组装效率比较。采用单因素方差分析差异。P<0.05认为具有显著性差异(***P<0.001,**P<0.01,*P<0.05)Fig.1 Assembly of 5 DNA fragments using ExoCET(a)Schematic diagram for the assembly.(b)EcoRⅠdigestion map for assembled plamids.The right side of the gel imagines is the theoretical map for the restriction digestion.1-24:Gibson+RecET,and 25-48:T4pol+RecET assembly.(c)Comparison of efficiencies for the assembly.Significance was analyzed by one-way ANOVA,and P<0.05 was considered statistically significant(***P<0.001,**P<0.01,and*P<0.05)
2.2 单拷贝BAC载体更适合高AT含量基因的克隆
为了测试ExoCET 组装更多高AT 含量DNA 片段的效率,分别将PCR 扩增的6 个、8 个和10 个3 kb MIT 9301基因组片段与1个2 kb pBR322载体,先在体外用Gibson体系进行处理,然后转化到表达RecET 重组酶的大肠杆菌中进行ExoCET 组装[图2(a)]。结果显示7 个片段和9 个片段均成功组装,而且菌落数均大于27 000 个/mL。在进行限制性酶切分析时,对相同培养时间并且相同体积的培养液进行质粒提取,然后溶解于相同体积的ddH2O后,取相同体积溶解好的质粒进行酶切。从胶图中可以看出,9片段组装质粒的浓度明显比7片段组装质粒的浓度低[图2(a)]。推测F7 和/或F8 片段中的DNA 对大肠杆菌有轻微毒性,影响了质粒拷贝数。11个片段的组装失败,可能是由于F9和/或F10片段对大肠杆菌也有毒性,导致正确重组的质粒无法在大肠杆菌中复制。接下来,尝试利用BAC载体与F1-F10片段进行组装,结果获得了大于2200个/mL的菌落数,正确率为100%[图2(b)]。

图2 ExoCET技术组装7~11个DNA片段(a)利用pBR322载体组装的示意图和EcoRⅠ酶切鉴定图:1~12为7个片段组装;13~24为9个片段组装;25~36为11个片段组装;胶图右侧从左到右依次为pBR322-amp-F1-6、pBR322-amp-F1-8、pBR322-amp-F1-10的酶切理论图谱(b)利用BAC载体组装11个片段的示意图和EcoRV酶切鉴定图,胶图右侧为酶切理论图谱Fig.2 Assembly of 7-11 fragments using ExoCET(a)Schematic diagrams and EcoRⅠrestriction analysis for the assembly using pBR322 vectors.1-12:7-piece assembly,13-24:9-piece assembly,and 25-36:11-piece assembly.The right side of the gel imagine is the theoretical map for the restriction digestion of pBR322-amp-F1-6,pBR322-amp-F1-8 and pBR322-amp-F1-10 from left to right.(b)Schematic diagrams and EcoRV restriction analysis for the 11-piece assembly using the BAC vector.The right side of the gel imagine is the theoretical map for the restriction digestion
使用pBR322质粒组装11个片段失败,而使用BAC 载体可以成功,推测这是由质粒拷贝数导致的。可能由于F9 和/或F10 片段的细胞毒性,当克隆载体选用拷贝数约为20的pBR322载体时,其表达量较高,毒性较强,超过细胞耐受水平,导致克隆失败。而选用单拷贝的BAC 载体时,其表达量控制在细胞可耐受的较低水平,所以能够克隆成功。此外,由于高AT含量DNA更容易发生呼吸作用(DNA breathing)形成单链DNA 构成的泡状结构,使基因表达相关的蛋白更容易结合在这些区域从而促进转录[14],导致毒性基因更容易表达而影响克隆。另一方面,如果pBR322 质粒携带了易发生呼吸作用的DNA 片段,其较高的拷贝数会占用宿主细胞大量的基因表达相关蛋白,进而影响宿主基因的转录导致细胞毒性,而单拷贝的BAC载体则可以避免此问题。
2.3 ExoCET 成功从原绿球藻MIT 9301 基因组上靶向抓取21~82 kb的大片段
为了测试ExoCET 抓取高AT 含量基因组大片段的可行性,利用PmeⅠ将MIT 9301 基因组切割成1.4~206 kb 的30 个DNA 片段[图3(a)],然后随机选取其中21 kb、25 kb、49 kb、50 kb、54 kb、65 kb、67 kb、82 kb 的8 个片段进行靶向抓取实验。首先在体外利用Gibson 体系对PmeⅠ酶切的MIT 9301 基因组和PCR 扩增的BAC 载体进行处理,然后转化到表达RecET 重组酶的大肠杆菌中进行ExoCET 靶向抓取[图3(b)]。BAC 载体两端带有目的基因组片段的两端的80 bp 同源臂。结果显示ExoCET 均能成功靶向抓取这8 个21~82 kb的基因组大片段[图3(c)~(j)],其中50 kb 片段的正确率最高,为87.5%;82 kb 片段的正确率最低,为4.2%。其余片段的正确率在11%~75%之间。克隆效率的总体趋势是片段越长克隆效率越低,这是由于基因组DNA 在制备过程中容易断裂,致使越大的目的DNA 片段在制备的基因组中含量越低,因此克隆效率也就越低。此外,长度接近片段的克隆效率也会存在较大差异,比如50 kb 片段的正确率为87.5%,而49 kb 片段的克隆正确率仅为23.5%,这可能是由于用于克隆49 kb 片段的同源臂经过核酸外切酶处理后产生的单链DNA 较容易产生二级结构,影响了载体和片段的退火。

图3 利用ExoCET-BAC策略进行直接基因克隆Fig.3 ExoCET-BAC strategy for direct gene cloning
2.4 ExoCET 从原绿球藻MIT 9301 基因组上一步同时抓取4个7~20 kb的大片段
系统开发基因组中的生物合成资源需要能够一步同时抓取多个片段的克隆技术。为了测试ExoCET 同时靶向抓取2 个片段的效率,随机选取了PmeⅠ酶切MIT 9301 基因组产生的2 组大小接近的片段,分别为10 kb、11 kb 和18 kb、22 kb 片段。首先将PmeⅠ酶切的MIT 9301 基因组和PCR扩增的2 个BAC 载体混合(其中2 个BAC 载体两端分别含有2 个对应目的片段两端的80 bp 同源臂),并利用Gibson 体系在体外进行处理,然后转化到表达RecET 重组酶的大肠杆菌中进行ExoCET靶向抓取[图4(a)]。结果显示ExoCET 均能实现上述两组片段的同时靶向抓取。从10 kb、11 kb 组筛选平板上随机挑取24 个菌落进行限制性酶切分析,发现其中3 个含有10 kb片段,10个含有11 kb片段[图4(b)]。从18 kb、22 kb 组筛选平板上随机挑取12 个菌落进行限制性酶切分析,发现其中1 个含有18 kb 片段,6 个含有22 kb 片段[图4(c)]。每组中两个大小接近片段的克隆效率差异较大,具体原因有待进一步解析。

图4 利用ExoCET-BAC策略进行两个片段一步抓取(a)一步抓取两个片段示意图;(b)直接克隆10 kb、11 kb片段EcoRⅠ酶切图(胶图右侧分别为直接克隆10 kb和11 kb基因组大片段的酶切理论图谱);(c)直接克隆18 kb、22 kb片段EcoRⅠ酶切图(胶图右侧从左到右依次为直接克隆22 kb和18 kb基因组大片段的酶切理论图谱)Fig.4 ExoCET-BAC strategy for capturing two DNA fragments simultaneously(a)Schematic diagram for the strategy.(b)EcoRⅠrestriction map for the direct cloning of 10 kb and 11 kb fragments.The right side of the gel imagine is the theoretical restriction map for the direct cloning of 10 kb and 11 kb fragments.(c)EcoRⅠrestriction map for the direct cloning of 18 kb and 22 kb fragments.The right side of the gel imagine is the theoretical restriction map for the direct cloning of 18 kb and 22 kb fragments from left to right
接下来,进一步测试了ExoCET 同时靶向抓取4 个片段的效率,此时随机选取了PmeⅠ酶切MIT 9301基因组产生的大小差异较大的4个片段,分别为7 kb、12 kb、18 kb和20 kb。首先将PmeⅠ酶切的MIT 9301 基因组和PCR 扩增的4 个BAC 载体混合(其中4个BAC载体两端分别含有4个对应目的片段两端的80 bp 同源臂),并利用Gibson 体系在体外进行处理,然后转化到表达RecET 重组酶的大肠杆菌中进行ExoCET 靶向抓取[图5(a)]。结果显示ExoCET 能实现上述4 个片段的同时靶向抓取。从筛选平板上随机挑取12 个菌落进行限制性酶切分析,发现其中2 个含有7 kb 片段,3 个含有12 kb 片段,1 个含有18 kb 片段,1 个含有20 kb 片段[图5(b)]。
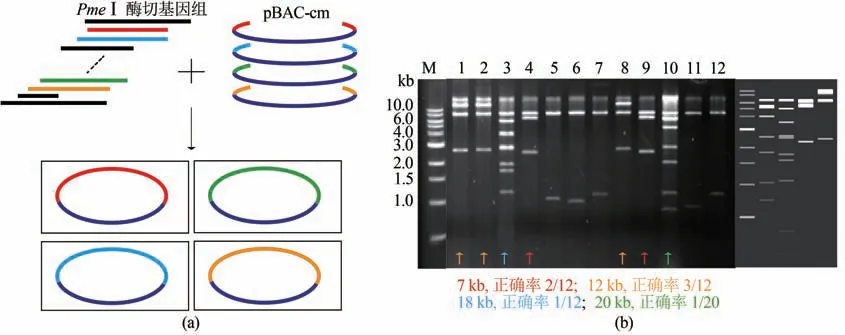
图5 利用ExoCET-BAC策略一步抓取4个片段(a)一步抓取4个片段示意图;(b)直接克隆7 kb、12 kb、18 kb和20 kb的EcoRⅠ酶切结果图(胶图右侧从左到右依次为直接克隆20 kb、18 kb、7 kb和12 kb基因组大片段的酶切理论图谱)Fig.5 ExoCET-BAC strategy for capturing four DNA fragments simultaneously(a)Schematic diagram for the strategy.(b)EcoRⅠrestriction map for the direct cloning of 7 kb,12 kb,18 kb and 20 kb fragments.The right side of the gel imagine is the theoretical restriction map for the direct cloning of 20 kb、18 kb、7 kb and 12 kb fragments from left to right
还测试了>22 kb 片段的多片段同时靶向抓取,但是均失败了,因此以目前的方法ExoCET 只能实现4个小于20 kb片段的同时靶向抓取。
3 结论
根据NCBI 2021 年6 月公布的基因组数据,已测序的327 130 个原核生物基因组中,有99 340 个(30.36%)微生物的基因组AT 含量≥60%。已测序的16 037个真核生物基因组中,有5274个(32.89%)真核生物基因组AT 含量≥60%。开发这些生物基因组中的生物合成资源需要更通用和便利的克隆技术。虽然已经有很多的直接克隆和多片段组装方法可以对DNA 大片段进行多片段组装和直接克隆,但是仍然有很多限制,导致高AT含量的DNA大片段克隆效率低下,或者无法进行克隆。本研究以AT 含量为69%原绿球藻基因组为对象,探讨了利用ExoCET 技术进行高AT 含量基因组大片段克隆的可行性,通过优化体外同源重组方式和载体拷贝数建立了ExoCET-BAC 策略。随机选取的16 个7~82 kb 的基因组大片段全部靶向抓取成功,其中对82 kb的DNA大片段进行靶向抓取的效率为4.2%。而且以100%的正确率组装了11 个3 kb 的DNA 片段。ExoCET-BAC 技术还能从原绿球藻基因组上一步同时抓取4 个7~20 kb 的基因组大片段,进一步证明了该技术的高效率。
最近,David J.Newman和Gordon M.Cragg对1981~2019 年所有FDA 批准的小分子药物的分析显示,这些化合物中有34%是天然产物或天然产物衍生物,其中相当一部分是由微生物产生的[27]。目前次级代谢产物研究最深入的微生物包括放线菌、黏细菌等[28]的基因组AT 含量均≤40%。链球菌、葡萄球菌和梭菌等高AT 含量微生物也含有丰富的有待开发的次级代谢产物生物合成基因簇[16,29]。ExoCET-BAC 技术将为这些生物合成资源的开发提供新手段。
随着合成生物学的不断发展,对生物基因组进行设计合成越来越受到人们关注[30-35],该研究建立的ExoCET-BAC 技术不仅有利于高AT 含量生物基因组“自上而下”的功能解析,还有利于高AT含量生物基因组“自下而上”的从头合成、组装和替换,丰富了合成生物学的使能技术工具箱。
致谢:感谢山东省泰山学者项目tsqn201812008 和山东大学齐鲁青年学者项目对本工作的支持。
猜你喜欢
塔里木大学学报(2022年4期)2022-12-24
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
基层中医药(2022年4期)2022-07-22
中国计量大学学报(2022年2期)2022-07-18
汉字汉语研究(2021年2期)2021-08-30
江西农业学报(2021年4期)2021-04-20
科技与创新(2020年12期)2020-11-28
三农资讯半月报(2020年11期)2020-06-21
汉字汉语研究(2019年2期)2019-08-27
