
ATP敏感性钾通道型先天性高胰岛素血症患儿临床及遗传学特征
2022-02-28 02:46惠培培徐子迪张琳曾俏刘敏闫洁吴玉筠桑艳梅朱逞倪桂臣李荣敏王杰英
中华胰腺病杂志 2022年1期
惠培培 徐子迪 张琳 曾俏 刘敏 闫洁 吴玉筠 桑艳梅 朱逞 倪桂臣 李荣敏 王杰英
1徐州医科大学附属医院儿科,徐州 221000;2国家儿童医学中心,首都医科大学附属北京儿童医院内分泌遗传代谢科,北京 100045;3北京儿童医院保定医院内分泌科,保定市儿童医院,儿童呼吸消化疾病研究重点实验室,保定 071000
先天性高胰岛素血症(congenital hypein-sulinism, CHI)是一种遗传异质性疾病,是婴幼儿和儿童时期顽固性、持久性低血糖的主要原因,其特点是顽固性低血糖及与血糖水平不相称的高胰岛素血症。随着研究的不断进展,迄今已发现了至少14种基因突变与CHI有关,相应地构成13种遗传学类型[1-3]。ATP敏感性钾通道型先天性高胰岛素血症(adenosine triphosphate-sensitive potassium channel hyperinsulinism, KATP-HI)是CHI最主要和常见的类型,由编码磺脲受体 1(sulfonylurea receptor 1, SUR1)的ABCC8基因和编码内向整流钾通道蛋白6.2(inward rectifying potassium channel,Kir6.2)的KCNJ11 基因突变引起。本研究总结及分析45例中国KATP-HI患儿的临床特征及携带的致病基因,旨在提高临床医师对该类型CHI的认识。
资料与方法
一、研究对象
选取2002年2月至2018年12月间首都医科大学附属北京儿童医院内分泌科收治的45例经遗传学确诊为KATP-HI的患儿及其家系为研究对象。本研究经医院伦理委员会批准,所有患儿父母均签署知情同意书。
二、诊疗过程
入院后45例患儿均确诊为CHI。CHI的诊断标准[4]包括高胰岛素血症(血浆胰岛素>2 mU/L)、低脂肪酸血症(血浆游离脂肪酸<1.5 mmol/L)、低酮血症(血浆β-羟丁酸<2.0 mmol/L)、1 mg静脉胰高血糖素试验反应血糖变化>0.3 g/L。
39例患儿应用二氮嗪进行试验性治疗,以5 mg·kg-1·d-1为起始剂量,每日2~3次口服,根据患儿病情逐渐增加剂量,最大剂量为15 mg·kg-1·d-1,疗程7~10 d。同时加用氢氯噻嗪利尿1~2 mg·kg-1·d-1,每日2~3次口服,以防止二氮嗪导致的水钠潴留,并予10%氯化钾1~2 ml·kg-1·d-1,每日3次口服,以防止氢氯噻嗪导致的低钾血症。二氮嗪治疗有效的判断标准[3,5]:患儿禁食12~18 h后血糖水平仍可维持在70 mg/dl(3.9 mmol/L)以上,或者血糖水平降至50 mg/dl(2.8 mmol/L)之前出现高酮血症(血浆β-羟丁酸>2.0 mmol/L)。如果应用二氮嗪最大剂量治疗5 d仍不能满足上述标准,则考虑为二氮嗪治疗无效[6]。二氮嗪治疗无效或疗效不确定的患儿中,18例进一步应用奥曲肽治疗,以5 μg·kg-1·d-1为初始剂量,间隔6 h或8 h皮下注射1次,根据患儿病情逐渐增加剂量,最大推荐剂量为20 μg·kg-1·d-1。奥曲肽疗效的判断标准同二氮嗪。部分内科治疗无效的患儿,通过胰腺切除术进一步控制血糖水平。
三、遗传学分析
1.血液DNA提取:留取患儿及其父母的乙二胺四乙酸钠抗凝静脉血3 ml,使用BloodGen Midi Kit(CWBIO,中国) 提取血中全基因组DNA,操作按照试剂盒说明书进行。
2.二代测序:采用IDT公司xGen Exome Research Panel v1.0捕获芯片。首先进行文库制备。(1)基因组片段化,Cavoris仪打断DNA片段至200 bp左右;(2)末端补平修复,片段化DNA经Klenow Fragment、T4 DNA polymerase和T4 PNK携带进行补平修复;(3)3′端腺苷化,应用聚合酶体系在补平修复产物的3′末端加上A碱基;(4)加接头,应用T4 DNA连接酶反应体系,在Thermo mixer中室温反应一定时间将dadpter连接到加“A”的产物;(5)扩增,连接产物经4~6轮PCR扩增;(6)杂交,PCR扩增产物与探针混于杂交体系,置65℃杂交60~68 h;(7)洗涤磁珠和洗脱DNA,杂交样本加入链霉素磁珠,孵育后用洗脱液洗脱;(8)洗脱产物扩增,洗脱产物经10轮LM-PCR扩增。然后进行Illumina平台测序。扩增产物经Illumina hisep xten平台标准化测序操作获取图像原始数据,通过Illumina官方baseball分析软件BclToFastq进行数据分析,并对突变假阳性进行过滤。
3.一代测序(Sanger法)验证:二代测序发现患儿携带ABCC8或KCNJ11基因突变,根据基因所验证位点序列设计引物,采用PCR方法进行扩增,用ABI 3730XL测序仪测序,测序引物采用原PCR引物;采用DNASTAR软件进行基因序列分析和比对。参照单核苷酸多态性数据库(dsSNP)和千人组基因库排除非致病性变异,并通过搜索在线人类孟德尔遗传数据库(OMIM)、人类基因突变数据库(The Human Gene Mutation Database)、NCBI数据库等明确该突变是否曾经报道过。根据人类基因组变异软件Provean、SIFT预测基因突变对蛋白结构的影响。
四、随访情况
对45例患儿进行6个月~10年的长期随访,主要随访内容包括患儿药物治疗、药物不良反应、术后转归、低血糖发作、空腹血糖及餐后2 h血糖、自行缓解等情况。CHI自行缓解的判断标准[7]:采用二氮嗪治疗的患儿停止口服二氮嗪至少5 d,空腹18~48 h(依年龄而定),血糖水平可维持在70 mg/dl(3.88 mmol/L),或者出现高酮血症(血浆β-羟丁酸>2.0 mmol/L)。
结 果
一、临床特征
45例KATP-HI患儿中男性26例,女性19例。出生体重1.8~5.0 kg,其中正常体重儿22例(48.9%),巨大儿22例(48.9%),低出生体重儿1例。发病年龄为出生当日~ 2岁,其中新生儿期发病34例(75.6%),1~6个月发病8例,>6个月发病3例。21例首发症状为抽搐,13例为反应弱,2例为多汗,1例为肌张力低,1例为面色青紫,7例无明显临床症状。1例患儿有低血糖家族史,其姐姐5岁时出现低血糖症状,后诊断为CHI。
二、诊疗效果
39例应用二氮嗪治疗的KATP-HI患儿中,12例(30.8%)治疗有效,16例(41.0%)治疗无效,另外11例患儿因二氮嗪治疗出现严重不良反应(血小板降低、严重的水钠潴留、不能耐受的胃肠道反应等)或家庭因素而终止二氮嗪治疗,故对二氮嗪疗效不明确。
二氮嗪治疗无效或疗效不确定而奥曲肽治疗应用的18例患儿,其中13例(72.2%)治疗有效,3例(16.7%)治疗无效,2例疗效不明确。
内科治疗无效的10例患儿行手术治疗,其中8例术前行18氟-左旋多巴正电子发射计算机断层扫描(18F-DOPA-PET)确定为局灶型病变的患儿行胰腺部分切除术;2例术前未行18F-DOPA PET扫描的患儿行次全胰腺切除术,术后病理检查结果显示1例为弥散型病变,1例为局灶型病变。
三、遗传学分析结果
45例KATP-HI患儿共发现52种ABCC8突变及2种KCNJ11突变,且经蛋白结构预测均可能有害或生物学危害性较高。10例携带ABCC8复合杂合突变(表1),34例只携带ABCC8或KCNJ11单基因突变(表2);1例男性新生儿出生后1 d发病,出生体重3.8 kg,血糖2.2 mmol/L,同时携带ABCC8基因(c.1775T>G)和KCNJ11基因(c.374T>C)突变,二氮嗪治疗有效。
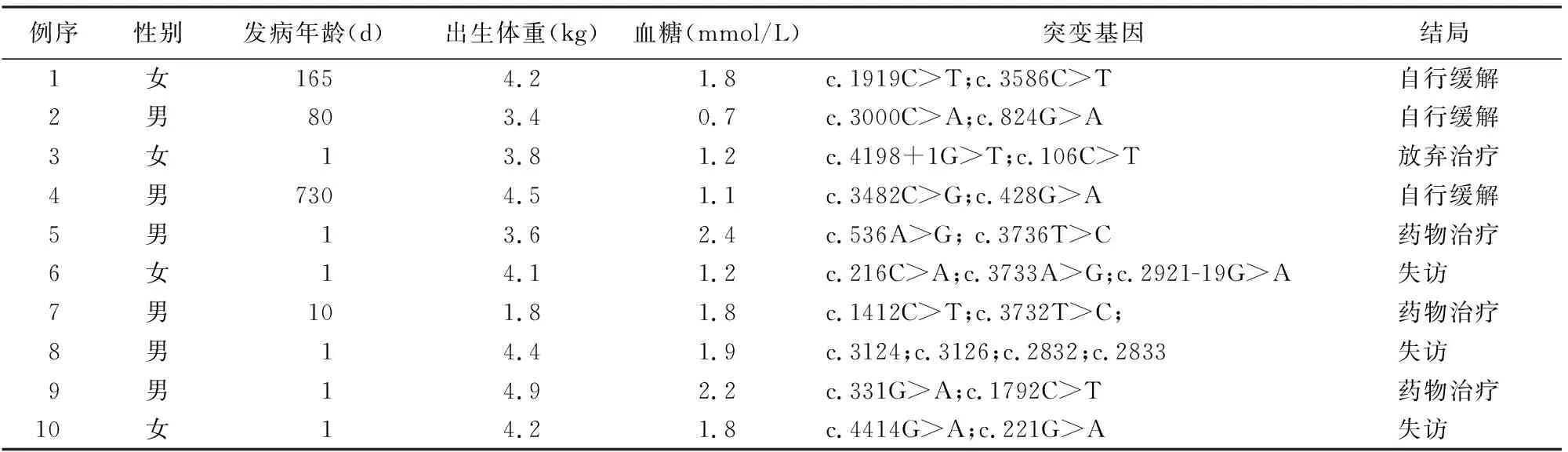
表1 10例ABCC8复合杂合突变的KATP-HI患儿临床特征及预后
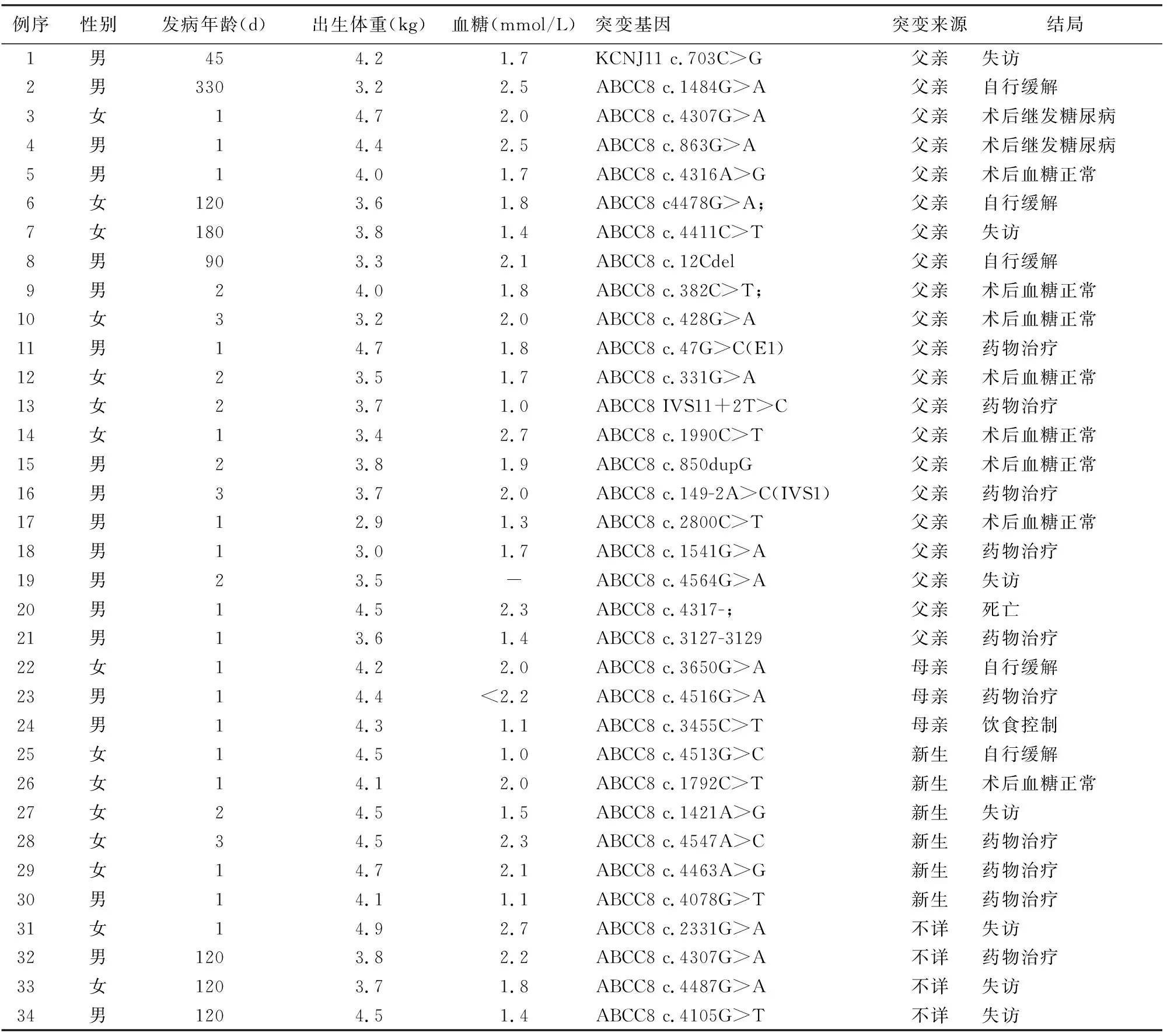
表2 34例ABCC8或KCNJ11基因单突变的KATP-HI患儿临床特征及预后
34例携带单个基因突变的患儿中21例突变来自于父亲,其中18例应用二氮嗪治疗,1例有效,7例无效,10例疗效不明确;3例突变来自于母亲,其中2例应用二氮嗪治疗,均有效,1例饮食控制血糖正常;新生突变6例,均应用二氮嗪治疗,3例有效,3例无效;4例突变来源不详。
四、随访结果
经长期随访,45例KATP-HI患儿中13例继续药物(二氮嗪或奥曲肽)治疗;8例自行缓解,缓解时间6个月~2岁,均为ABCC8突变,其中6例为单基因突变,2例为复合杂合突变;8例行胰腺部分切除治疗的KATP-HI患儿,术后血糖恢复正常;2例行次全胰腺切除术的患儿分别于术后第3年、第4年并发1型糖尿病;1例频繁喂养下未出现频繁低血糖;1例放弃治疗;11例失访;1例因呼吸道感染死亡。
讨 论
KATP-HI是CHI最常见类型,是由于ABCC8及KCNJ11基因缺失性突变导致的。KATP-HI患儿发病较早,多见于新生儿期。既往文献报道,新生儿期发病者约占83%[8],本研究45例KATP-HI患儿中新生儿期发病者占75.6%,与文献报道基本一致。KATP-HI患儿中巨大儿占比较高,本研究的巨大儿约占48.9%,与文献报道的45.3%接近[8]。
二氮嗪是CHI的一线治疗药物,该药为钾通道开放剂,能与KATP敏感性钾通道的SUR1亚单位结合,使钾通道处于开放状态,从而抑制胰岛素的分泌。研究资料显示,二氮嗪治疗对多数KATP-HI患儿无效(62.0%~86.8%),机制与KATP-HI编码钾通道的基因出现不同程度的功能缺失性突变有关[8-9]。本组39例患儿曾予二氮嗪治疗,无效率高达41.0%,稍低于文献报道,考虑与部分患儿疗效尚未明确有关。绝大多数二氮嗪治疗无效的CHI患儿奥曲肽治疗效果较好。本组对二氮嗪无效或疗效不确定的18例患儿进一步选择奥曲肽治疗,有效率高达72.2%,与既往文献报道一致。
对内科治疗无效的CHI患者可考虑行胰腺切除术。国外文献建议的手术指征为内科治疗无效的CHI患儿或经18F-DOPA-PET确定为局灶型病变的患儿[10]。2019年美国的一项研究报道246例行胰腺部分切除术治疗的局灶型CHI患儿,97%术后获得治愈,仅7例仍需要通过药物控制低血糖。本组8例行胰腺部分切除术的患儿均获得治愈,说明局灶型CHI行胰腺部分切除术有较高的临床治愈率。次全胰腺切除术的常见并发症主要有反复低血糖、糖尿病和外分泌功能不全,本组2例行次全胰腺切除术的患儿均继发糖尿病,表明术前明确病理类型对手术方式的选择及预后评估极为重要。
研究显示, CHI 患儿随着年龄的增长及其对胰岛素需要量的增加,其低血糖症状可有所减轻,部分患儿可自愈。国外研究报道,约48%CHI患儿的低血糖可自行缓解,且自行缓解的时间跨度为3个月~8岁,自行缓解的患儿中约15%是KATP-HI,4%分别为其他类型基因突变[11]。本组8例患儿自行缓解,时间跨度为6个月~2岁。说明应适当延长 CHI 患者的随访时间,对二氮嗪治疗无效的 CHI 患者应谨慎选择手术治疗,从而尽可能减少手术并发症的发生。
随着研究的进展,迄今已发现448种ABCC8突变,66种KCNJ11基因突变[12]。携带ABCC8基因复合杂合突变的KATP-HI患儿发病时间多较早,组织学类型均为弥散型病变[1],少数患儿对二氮嗪治疗有效,绝大多数对奥曲肽治疗有效[13-14]。本组10例携带复合杂合突变的KATP-HI患儿中9例曾应用二氮嗪治疗,仅3例有效,5例无效患儿进一步应用奥曲肽治疗,其中4例有效。提示携带ABCC8复合杂合突变的CHI患者多对二氮嗪无效,与文献报道一致[13,15]。研究发现,94%携带ABCC8或KCNJ11基因单一杂合隐性突变患儿的胰腺组织学病变为局灶型,如果突变遗传自父亲,则病变类型为局灶型的可能性上升至98%[16]。隐性遗传的KATP-HI患儿临床低血糖症状多较重,且多对二氮嗪治疗无反应。携带ABCC8或KCNJ11基因单一杂合显性遗传患儿的胰腺组织学类型多为弥散型,临床症状较隐性遗传者轻,且绝大多数二氮嗪治疗有效,这与显性遗传的ATP敏感性钾通道有部分功能相关[16]。本组7例携带ABCC8父系遗传的患儿均为局灶型,均对二氮嗪治疗无效,后行胰腺部分切除术,术后随访,在无药物治疗下血糖均正常。根据患儿的临床表现、二氮嗪治疗情况及组织学类型考虑这7例患儿为常染色体隐性遗传,但因未对胰腺病灶进行微卫星标记分析,暂不能证实母系11p15.1染色体等位基因的缺失。
既往研究发现,母系遗传的携带单个ABCC8或KCNJ11基因突变的CHI患者中巨大儿多见,主要于婴儿期发病,临床症状较父系遗传的隐性突变的CHI患儿轻,多对二氮嗪治疗有效,对二氮嗪试验性治疗无反应者可给予奥曲肽,均治疗有效,迄今尚未见此类患者行手术治疗。文献报道此类患者遗传方式多为常染色体显性遗传,组织学类型多为弥散型[16-17]。本研究的3例母系遗传患儿均为巨大儿,其中2例应用二氮嗪治疗,均有效,1例饮食控制血糖正常,符合文献报道的母系遗传KATP-HI的临床特征。
新生突变所致的KATP-HI遗传方式为常染色体显性遗传,组织学类型多为弥散型,部分对二氮嗪治疗无效[13,17]。本研究6例新生突变,均应用二氮嗪进行治疗,其中3例有效,3例无效。无效患儿中1例曾行18F-DOPA PET,结果显示为局灶型,后行胰腺部分切除术治愈,1例予以奥曲肽治疗后血糖稳定,1例失访,提示新生突变所致的KATP-HI临床症状轻重不一,局灶型CHI可能有着更为复杂的遗传学机制。
Rozenkova等[18]曾报道1例同时携带ABBC8(p.Tyr1293Asp)及KCNJ11(p.Arg50Trp)突变的患儿,突变基因分别来自其父亲和母亲,足月顺产,出生体重4 kg,生后第1天发病,临床症状较轻,对二氮嗪无效但对奥曲肽治疗有效,并于2岁半时自行缓解。本研究也发现1例同时携带ABBC8(p.L592R)及KCNJ11(p.I125T)突变的患儿,足月顺产,出生体重3.8 kg,出生后第1天发病,对二氮嗪治疗有效。患儿现5岁,仍口服二氮嗪治疗,血糖可维持在正常范围内,提示同时携带ABBC8及KCNJ11基因突变的CHI患者发病均较早,临床症状较轻,且均对药物治疗有效。
综上所述,KATP-HI患者发病多较早,临床症状轻重不一,多数对二氮嗪的治疗无效,而奥曲肽的疗效较好。局灶型病变患者行胰腺部分切除术治疗具有较高的治愈率,次全胰腺切除术有继发糖尿病的风险,故术前明确患者的胰腺组织学类型极为重要。部分KATP-HI患儿有自行缓解的可能,故可以适当延长随访时间,谨慎选择手术治疗。携带ABCC8基因复合杂合突变的KATP-HI患儿发病时间早,均为弥散型病变。携带ABCC8或KCNJ11单个基因突变的父系遗传的KATP-HI患儿,隐性遗传者的胰腺组织学类型多为局灶型,临床低血糖症状较重,多对二氮嗪治疗无反应;显性遗传者的胰腺组织学类型多为弥散型,临床症状较轻,绝大多数对二氮嗪治疗有效,这可能与显性遗传的ATP敏感的钾离子通道有部分功能相关。
利益冲突所有作者声明无利益冲突
作者贡献声明惠培培:论文撰写、统计分析;徐子迪、张琳、 曾俏:采集数据、数据整理;刘敏、闫洁、吴玉筠:分析数据;朱逞、倪桂臣:获取材料支持;桑艳梅:研究指导、论文修改
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
中国听力语言康复科学杂志(2021年6期)2021-12-21
健康之家(2021年6期)2021-09-08
科学导报(2021年29期)2021-06-03
人人健康(2020年4期)2020-05-25
科海故事博览·下旬刊(2019年6期)2019-04-16
中学生理科应试(2017年6期)2017-09-27
恋爱婚姻家庭·养生版(2016年11期)2016-11-03
中国民族民间医药·下半月(2011年10期)2011-12-27
