
组氨酸6.52和酪氨酸7.43位点突变对 μ阿片受体下游信号通路的影响
2022-01-08 10:30田祥云刘晓倩苏瑞斌
中国药理学与毒理学杂志 2021年12期
田祥云,孙 毅,邵 帅,刘晓倩,谭 博,苏瑞斌
(1.南京中医药大学,江苏 南京 210000;2.军事科学院军事医学研究院毒物药物研究所,抗毒药物与毒理学国家重点实验室,北京 100850)
阿片类生物碱及相关药物是治疗急慢性疼痛最有效的镇痛药,其中吗啡和可待因是主要的活性阿片类生物碱。早期研究表明,阿片类药物及其衍生物作用于中枢神经系统的μ阿片受体(μ-opioid receptor,MOR)后,主要通过抑制性G蛋白(Gi)与细胞效应因子相互作用来传递镇静和镇痛等[1-2]。近年来,人们对β-制动蛋白分子的认识不断加深,提出了β-制动蛋白可以作为G蛋白偶联受体(G protein-coupled receptors,GPCR)的信号转导器,独立于G蛋白介导下游信号通路的传递的理论,这与以往认定的β-制动蛋白是G蛋白信号通路的负性调节因子观点有所不同。GPCR结构解析的进一步研究表明,受体的激活构象是动态可变的,与不同配体结合会呈现出不同的激动态结构,从而不同程度地招募G蛋白和β-制动蛋白,触发不同的信号通路,最终引起千差万别的效应[3-4]。
随着人们对MOR结构解析研究的深入,如2012年解析的MOR-β-富纳曲胺(funaltrexamine,FNA)(拮抗剂)复合物结构[5]、2015年解析的MOR-桥联吡咯烷吗啡喃(bridged pyrrolidinomor⁃phinan,BU72)(激动剂)复合物结构[2]和2018年解析的 MOR-DAMGO-Gi蛋白复合物结构[6],均提示了不同配体与MOR相互作用后,MOR发生不同的构象变化,最终形成不同的稳定构象[2-4]。Smith等[7]提出“三元复合物”模型从结构上解释受体、配体和信号转导蛋白之间的相互作用。当配体与受体结合后,受体会发生构象转变,形成中间构象状态,而这一状态决定了受体是结合G蛋白还是结合β-制动蛋白形成最终稳定状态。结构解析研究显示,受体中的某些基元如NPxxY,可能与β-制动蛋白的招募有关[8],而某些基元如脯氨酸5.50-异亮氨酸3.40-苯丙氨酸6.44三联体,可能与G蛋白信号通路相关[2]。本研究以吗啡和DAMGO为例,采用计算机模拟方法预测影响其与MOR结合的氨基酸位点,将相应位点突变后,通过G蛋白依赖性信号通路和β-制动蛋白依赖性信号通路的激活程度来分析其中的关键氨基酸位点。
1 材料与方法
1.1 药品、试剂和仪器
芬太尼(军事医学研究院毒物药物研究所合成,纯度99%);盐酸吗啡(国药准字H63020013,青海制药厂有限公司,批号:20020201);DAMGO(批号:1171,美国Tocris Bioscience公司);纳曲吲哚(naltrindole hydrochloride,批号:14A/204708,美国Tocris Bioscience公司);胎牛血清、DMEM培养基和Opti-MEM培养基和不依赖CO2的培养基(美国Gibco公司);定点突变试剂盒(南京诺唯赞生物科技股份有限公司);无内毒素质粒大提试剂盒(德国QIAGEN公司);DNA纯化回收试剂盒、2×Pfu PCR MasterMix和DNA Marker 2000(北京天根生物技术公司);DH5α感受态细胞(北京博迈德基因技术有限公司),琼脂糖(西班牙Biowest Agarose公司);胰蛋白胨和酵母提取物(英国Oxoid公司);NaCl(西陇科学股份有限公司);氨苄青霉素、卡那霉素和毛喉素(forskolin,FOR)(美国Sigma公司);Q5热启动超保真2×Master Mix(美国NEB公司);NheⅠ限制性内切酶、SacⅠ限制性内切酶、EcoRⅠ限制性内切酶、T4 DNA连接酶、10×T4 DNA连接酶缓冲液、反转录cDNA合成试剂盒、SYBR Green/ROX qPCR试剂盒和Lipofectamine®3000转染试剂(美国Thermo Scientific公司);M5 Gelred Plus核酸染料(北京聚合美生物科技有限公司);SNAP-Lumi4-Tb、5×Tag-lite标记缓冲液和Taglite MOR荧光标记配体(美国Cisbio公司);兔抗人MOR多克隆抗体(美国Minipore公司);辣根过氧化物酶(HRP)标记的山羊抗兔多克隆抗体、HRP标记的山羊抗小鼠多克隆lgG和小鼠抗大鼠GAPDH单克隆抗体(北京普利采基因技术公司);小鼠抗人β-制动蛋白单克隆抗体(美国CST公司);DMSO(美国MP Biomedical公司);pGloSensorTM-22F cAMP 质粒(a-f)、GloSensorTMcAMP Reagent、Nano⁃BiT®PPI MCS Starter System和Nano-Glo®活细胞分析系统(美国Promega公司);重组质粒构建以及实时定量PCR所需引物由生工生物工程(上海)股份有限公司合成。
基因扩增仪(北京东胜创新生物科技有限公司);电泳仪和水平电泳槽(北京市六一仪器厂);紫外透视仪(海门市其林贝尔仪器制造有限公司);多功能成像仪(美国Kodak公司);紫外可见分光光度计(美国Unico公司);倒置显微镜(重庆光电仪器有限公司);多功能酶标仪(美国Perkin Elmer公司);CO2培养箱(美国Thermo Scientific公司);离心机(德国Eppendorf公司);实时荧光定量PCR仪器(美国Applied Bionsystem公司)。
1.2 细胞和细胞培养
HEK 293T细胞(中国典型培养物保藏中心细胞库),用含10%胎牛血清的DMEM培养液在37℃,5% CO2恒温培养箱中培养。
1.3 计算机模拟预测MOR与配体相互作用的关键位点
本实验室与复旦大学付伟教授团队合作进行DAMGO和吗啡与MOR的分子对接。使用Discov⁃ery Studio 3.5软件中同源模建模块,以小鼠激动态MOR的X射线晶体结构(PDB编码:5C1M)[2]为模板建立人源激动态MOR三维结构模型,原MOR晶体结构中的所有水分子均保留;使用Maestro 3.5程序中的Glide Docking模块将DAMGO和吗啡对接到MOR的结合位点,从而得到化合物与MOR的分子对接图。
1.4 SNAP-MOR突变体质粒的构建
以本实验室谭博构建的SNAP-MOR质粒为模板设计引物,将质粒进行反向PCR扩增并引入突变碱基,引物序列见表1。反应体系:2×Max缓冲液12.5 μL,正向引物(10 μmol·L-1)1.0 μL,反向引物(10 μmol·L-1)1.0 μL,dNTP 混合物(10 mmol·L-1)0.5 μL,Phanata Mix Super-Fidelity DNA 聚合酶0.5 μL,SNAP-MOR(500 mg·L-1)0.5 μL,双蒸水9.0 μL。反应过程:① 95℃ 30 s,② 95℃ 15 s,③64~68.5℃ 15 s,④ 72℃ 5 min,⑤ 72℃ 5 min,其中过程②~④循环33次,退火采用程序升温,每个循环增加0.1℃;其中的退火温度:Y7.43F为65.5~68.5℃;H6.52L,Y7.43A和Y7.43S为61~64℃;H6.52D和H6.52W为64~67℃。

Tab.1Sequence of primers for μ-opioid receptor(MOR)site mutation
扩增产物经琼脂糖凝胶电泳分离后得到目的基因(6000 bp),用DNA纯化回收试剂盒将目标质粒纯化回收后经DpnⅠ消化,去除甲基化模板质粒,反应体系为DpnⅠ1 μL,扩增产物40 μL;反应条件为37℃恒温2 h;消化完成后将产物进行重组反应,反应体系为5×CE Ⅱ缓冲液 8 μL,DpnⅠ消化产物 2 μL,exnase Ⅱ4 μL,双蒸水26 μL;置37℃恒温反应30 min,冰水浴冷却5 min;将反应产物转化到DH5α感受态细胞中,接种到含氨苄青霉素抗性的LB固体培养基,37℃培养24 h后挑取若干单克隆,在扩大培养后提取重组质粒,进行目的基因的测序比对。
1.5 荧光标记配体结合实验
1.5.1 细胞分组处理和荧光强度检测
将SNAP-MOR和SNAP-MOR突变体质粒转染至HEK 293T细胞,待细胞贴壁伸展汇合度约80% 时,换含有 SNAP-Lumi4-Tb(100 nmol·L-1)工作液的培养基,置37℃,5% CO2培养箱中孵育1 h。弃SNAP-Lumi4-Tb工作液,用1×Tag-lite标记缓冲液清洗细胞后加入0.02%乙二胺四乙酸溶液消化细胞,经离心、重悬后制成单细胞悬液,并调整细胞密度为5×108L-1。使用荧光标记配体(即带有红色荧光基因的纳曲酮衍生物)作为结合分子进行荧光标记配体结合实验。在384孔板中每孔加入10 μL单细胞悬液,设如下组别:①荧光配体总结合组〔每孔加5 μL 1×Tag-lite标记缓冲液、5 μL梯度浓度的荧光标记配体(终浓度0.1,0.2,0.4,0.8,1.6,3.2,6.3,12.5,25.0,50.0和100.0 nmol·L-1)〕;② 荧光配体非特异结合组〔每孔加5 μL 10 μmol·L-1的纳曲吲哚、5 μL梯度浓度的荧光配体(终浓度同①)〕;③ 药物竞争结合组〔每孔加5 μL 32 nmol·L-1的荧光标记配体、5 μL梯度浓度的DAMGO或吗啡(终浓度均为10-14~10-4mol·L-1)〕,每组设2复孔。加样完成后,将384孔板贴膜封闭,微震混匀,避光孵育3 h,用多功能酶标仪检测在激发光340 nm,发射光665和615 nm波长下的荧光强度(fluorescence intensity,FI),以均相时间分辨荧光(homoge⁃neous time resolved fluorescence,HTRF)比值反映荧光标记配体与MOR的结合程度,HTRF比值=FI665nm/FI615nm×104。实验重复3次。
因此,选择适合香菇优良品种,综合考虑品种特性、市场需求和当地气候特点[7],结合区域气候、地形、人力等基本情况,加强并提高技术管理形成规范化的模式和技术规程,对于引进菌类品种的栽培成功非常重要。跟进当地气候变化情况,因地制宜及时进行技术、管理调整,多参考前人成功事例,总结失败与成功的经验,是产业落地稳定可持续发展的重要步骤。
1.5.2 统计学分析
1.6 GloSensor cAMP生物传感器测定cAMP含量
1.6.1 细胞分组处理和cAMP含量检测
将SNAP-MOR和SNAP-MOR突变体质粒分别与pGloSensorTM-22F质粒转染至HEK 293T细胞[9],在转染16~18 h后,取密度为1.5×108L-1的单细胞悬液100 μL接种到96孔板,12 h后弃培养液,换成平衡培养基(含88%不依赖CO2的培养基、10% FBS和2% GloSensor cAMP试剂原液)在37℃,5% CO2培养箱中孵育1 h,孵育完成后,进行加药处理,多功能酶标仪测定400~700 nm范围内的化学发光强度(luminescence intensity,LI)。细胞分为空白组(每孔90 μL平衡培养基);FOR组(每孔99 μL平衡培养基,加入 1 μL FOR 10 μmol·L-1);药物处理组〔每孔90 μL平衡培养基,加入10 μL梯度浓度的DAMGO或吗啡(浓度为10-11~10-3mol·L-1),再每孔加入1 μL FOR 10 μmol·L-1〕,20 min后测定LI,以LI反映细胞内cAMP含量。每组设3复孔,计算药物作用后cAMP抑制百分率,以反映药物对MOR下游G蛋白依赖性信号通路的激活。cAMP抑制百分率(%)=〔(FOR组LI-空白组LI)-(药物处理组LI-空白组LI)〕/(FOR组LI-空白组LI)×100%。实验重复3次。
1.6.2 统计学分析
1.7 蛋白质相互作用分析体系测定配体激活MOR后对 β-制动蛋白2的募集
此分析系统需构建MOR-SmBiT和LgBiTARRB2表达载体。MOR-SmBiT表达载体以SNAP-MOR质粒为模板,正向引物:5’-CTAGC⁃TAGCATGGACAGCAGCGCG-3’,反向引物:5’-ATACGAGCTCTGGGCAACGGAGCAG-3’,在5’端引入NheⅠ限制性内切酶位点,3’端引入SacⅠ限制性内切酶位点;LgBiT-ARRB2表达载体以Flag-ARRB2质粒为模板,正向引物:5’-ATAC⁃GAGCTCCATGGGGGAGAAACC-3’,反向引物:5’-CTAGCTAGCCTAGCAGAGTTGATCATC-3’,在5’端引入SacⅠ限制性内切酶位点,3’端引入NheⅠ限制性内切酶位点。PCR扩增MOR片段,反应体系:Q5高保真热启动2×混合液12.5 μL,正向引物(10 μmol·L-1)1.25 μL,反向引物(10 μmol·L-1)1.25 μL,SNAP-MOR(500 mg·L-1)模板DNA 1.0 μL〔(SNAP-MOR(500 mg·L-1),Flag-ARRB2(869 mg·L-1)〕,双蒸水9.0 μL;反应过程:① 95℃ 1 min,② 95℃ 10 s,③ 65℃ 30 s,④ 72℃ 2 min,⑤ 72℃4 min,其中过程②~④循环30次。
PCR产物进行琼脂糖凝胶电泳,对目的条带1200和1400 bp左右的胶进行回收。将扩增片段与目的载体进行酶切和连接。片段酶切体系:10×FastDigest缓冲液 4 μL,NheⅠ限制性内切酶2 μL,SacⅠ限制性内切酶 2 μL,片段 DNA 5 μL,双蒸水27.0 μL;载体酶切体系同片段酶切体系,酶切条件为37℃水浴5 min。酶切产物进行琼脂糖凝胶电泳,确认产物条带位置无误后,回收酶切产物进行连接。连接体系:10×T4 DNA连接酶缓冲液2 μL,T4 DNA连接酶0.2 μL,线性载体DNA 3 μL,插入片段DNA 14 μL,双蒸水0.8 μL。连接条件:22℃孵育10 min,65℃热失活10 min。将连接产物转化到大肠杆菌DH5α感受态细胞中,从转化平板上挑取单克隆,利用菌液PCR的方法筛选阳性克隆,PCR反应体系:2×PCR 混合液 5 μL,正向引物(10 μmol·L-1)1.0 μL,反向引物(10 μmol·L-1)1.0 μL,菌液 2.0 μL,双蒸水1.0 μL;PCR反应过程同1.4。将PCR产物再次进行琼脂糖凝胶电泳,验证重组载体的构建,从筛选出的阳性克隆中抽提质粒进行测序。
1.7.2 细胞处理和检测
突变型MOR-SmBiT表达载体以MOR-SmBiT质粒为模板进行构建,方法同1.4。分别将MORSmBiT、MOR-SmBiT突变体质粒和LgBiT-ARRB2质粒共同转染至HEK 293T细胞[9],在转染16~18 h后,取密度为1.5×108L-1的单细胞悬液100 μL接种到96孔板,12 h后弃培养液,用Opti-MEM缓冲培养基在环境温度中平衡10 min,平衡完成后,每孔加入25 μL Nano-Glo活细胞试剂,将细胞分为空白组和DAMGO或吗啡处理组(每孔加入10 μL DAMGO或吗啡,分别为1.35×10-11~1.35×10-3mol·L-1),作用15 min后用多功能酶标仪测定400~700 nm范围内的LI。以LI指标反映药物作用后β-制动蛋白2的招募量,每组3复孔,计算药物作用后β-制动蛋白2的招募量。β-制动蛋白2的招募量=药物处理组LI-空白组LI。实验重复3次。
1.7.3 统计学分析
2 结果
2.1 DAMGO和吗啡与MOR相互作用关健位点
人源激动态MOR三维结构模型见图1A,DAMGO和吗啡与MOR的分子对接图确定了DAMGO和吗啡与MOR的结合模式(图1B和1C)。结果显示,DAMGO和吗啡的质子化氮均与MOR中D3.32的羧基氧原子形成稳定的离子键,Y7.43的羟基与D3.32的羧基形成氢键;吗啡结构中苯环上的羟基通过水桥与H6.52的咪唑侧链相互作用、与K5.39的羧基氧相互作用,从而形成较复杂的氢键网络。由此推断,H6.52和Y7.43参与DAMGO和吗啡与MOR的相互作用。
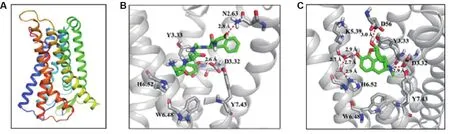
Fig.1 Three dimensional structure models of human dynamic μ-opioid receptor(MOR)(A)and details of binding modes of DAMGO(B)and morphine(C)with wild type MOR.
2.2 DAMGO和吗啡与野生型和各突变型MOR的竞争结合
荧光标记配体饱和结合曲线(图2A)显示,荧光标记配体与H6.52L,H6.52D和H6.52W突变型MOR特异结合的能力极弱,但可与Y7.43A,Y7.43F和Y7.43S突变型MOR特异结合,Kd值和Bmax值见表2。通常认为,荧光标记配体与突变型MOR结合的Kd值若处在野生型MOR的4倍Kd〔(13.1±1.5)nmol·L-1〕范围内则无明显差异。实验结果提示,Y7.43位点突变不改变荧光标记配体与受体的结合。
荧光标记竞争结合曲线(图2B和2C)显示,DAMGO、吗啡与Y7.43A,Y7.43F和Y7.43S突变型MOR的竞争结合曲线相对于野生型MOR无变化。分别将DAMGO和吗啡作用于Y7.43A,Y7.43F和Y7.43S突变型MOR后,与荧光标记配体竞争的Ki值与作用于野生型MOR的Ki值比较(表2),均处在野生型MOR的4倍Ki范围内,提示Y7.43位点突变不改变DAMGO和吗啡与MOR的亲和力。
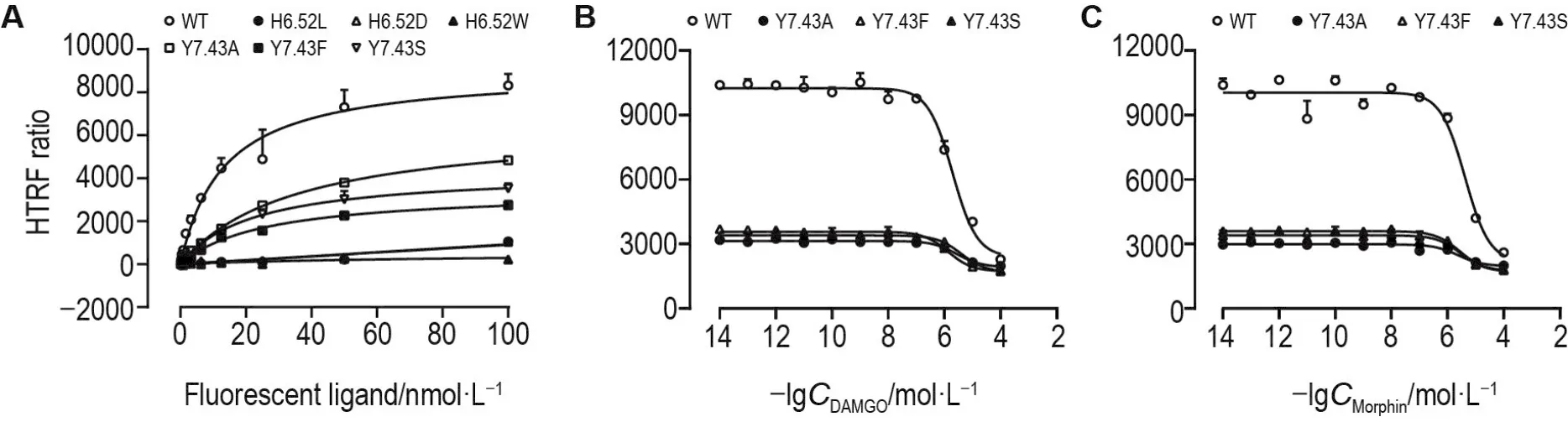
Fig.2 Site-mutations influence binding properties of fluorescent ligand with MOR.HEK293T cells were labeled in batch with SNAP-Lumi4-Tb and suspended in Tag-lite labeling buffer(TLB),then cells were divided into total binding,non-specific bind⁃ing and drug competitive binding groups.For total binding group,cells were incubated with fluorescent ligand(0.1-100 nmol·L-1)in TLB.Non-specific binding signal wells were incubated with 100 nmol·L-1naltrindole and fluorescent ligand(0.1-100 nmol·L-11).For drug competitive binding group,cells were incubated with DAMGO and morphine(10-14-10-4mol·L-1)in the presence of 8 nmol·L-1flu⁃orescent ligand.The fluorescence intensity(FI)at 665 nm and 615 nm was detected after 3 h incubation.A:fluorescent ligand satura⁃tion binding of wild-type(WT)and mutant MOR;B:homogeneous time-resolved fluorescence(HTRF)competition binding of fluores⁃cent ligands to DAMGO;C:HTRF competition binding of fluorescent ligands to morphine.The fluorescent ligand is a naltrexone deriva⁃tive with red fluorescent probe.HTRF ratio=FI665 nm/FI615 nm×104.±s,n=3.

Tab.2 Saturated binding parameters and competitive binding constants
2.3 配体对MOR下游腺苷酸(adenylic acid,AC)-环磷酸腺苷(cyclic adenosine monophosphate,cAMP)通路的激活
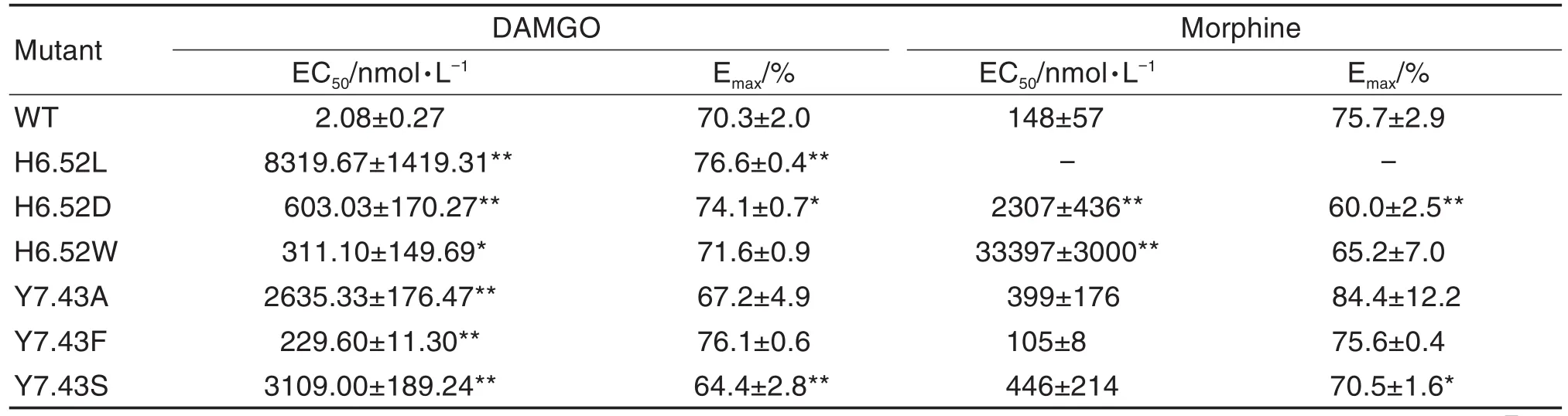
GloSensor cAMP实验通过测定cAMP含量变化来反映药物作用于MOR后对下游AC-cAMP通路的激活程度。DAMGO和吗啡作用于H6.52突变型MOR后,与其作用于野生型MOR相比,量效曲线均右移(图3A和3B)。H6.52L,H6.52D和H6.52W 3种突变型MOR均减弱了DAMGO和吗啡刺激下的AC-cAMP通路激活,其中H6.52L极大程度地减弱吗啡刺激下AC-cAMP通路的激活,由量效曲线计算得出的EC50和Emax值见表3。
Tab.3 Activation parameters of test agonists to stimulate MOR-mediated G-protein-dependent signaling
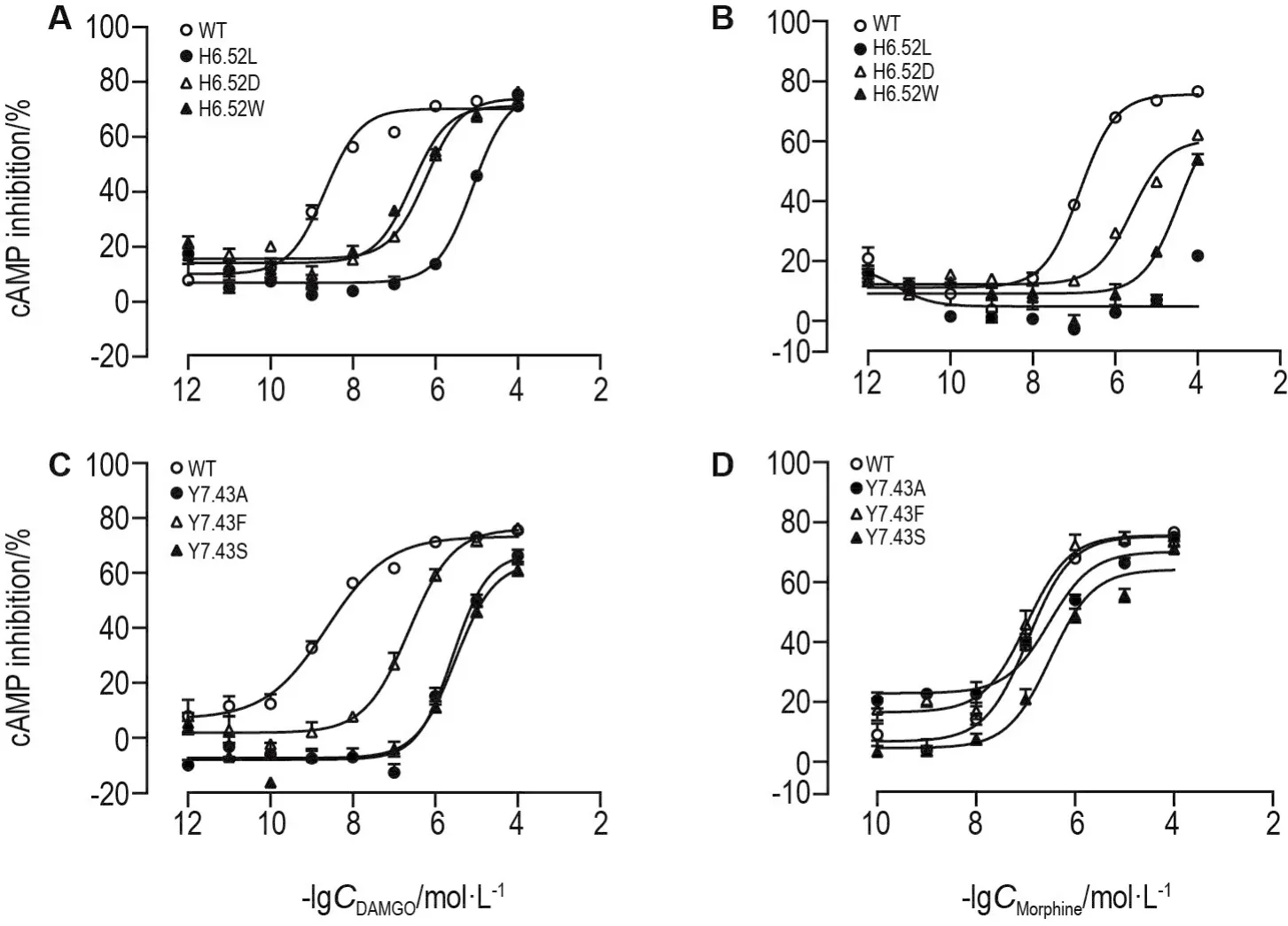
Fig.3 Effcet of DAMGO and morphine on forskolin-stimulated cAMP accumulation in H6.52 and Y7.43 mutant MOR.HEK293T cells were divided into blank,forskolin and drug groups.Drug groups were treated with DAMGO or morphine(10-12-10-4mol·L-1),forskolin and drug groups were treated with forskolin(10-5mol·L-1)sequentially for 20 min,then luminescence intensity(LI)was detected.cAMP inhibition(%)=(LI of forskolin group-LI of drug group)/(LI of forskolin group-LI of blank group)×100%.±s,n=3.
Y7.43A,Y7.43F和Y7.43S突变明显减弱DAMGO对MOR下游AC-cAMP通路的激活,其中Y7.43A和Y7.43S减弱MOR下游通路激活的程度强于Y7.43F(P<0.01);Y7.43突变后不改变吗啡对MOR下游AC-cAMP通路的激活(图3C和3D),Y7.43A,Y7.43F和Y7.43S 3种突变型对MOR下游AC-cAMP通路的激活程度基本一致,由量效曲线计算得出的EC50和Emax值见表3。
2.4 配体激活MOR下游 β-制动蛋白2的招募
利用NanoBit蛋白相互作用分析方法检测MOR和β-制动蛋白2的相互作用,反映MOR下游β-制动蛋白依赖性信号通路的激活程度。H6.52和Y7.43突变阻断了DAMGO和吗啡对MOR下游β-制动蛋白依赖性信号通路的激活。DAMGO作用于野生型MOR后会引起下游β-制动蛋白依赖性信号通路的激活,其EC50值为(1184±112)nmol·L-1,而H6.52和Y7.43突变后均大幅度减弱该通路的激活(图4A);吗啡作用于野生型MOR后未引起下游β-制动蛋白依赖性信号通路的激活,H6.52和Y7.43突变后该通路仍保持未激活状态(图4B)。
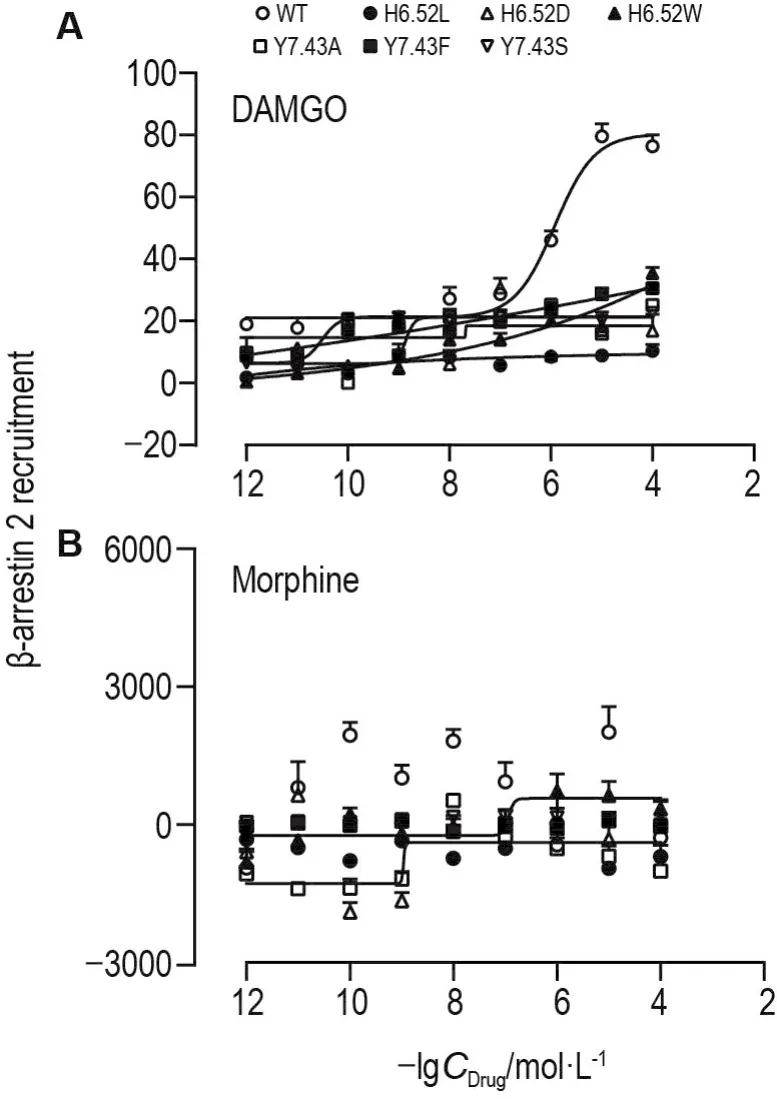
Fig.4 Functional activity of test agonists to stimulate MOR-mediated β-arrestin-dependent signaling.HEK293T cells were divided into blank group and drug groups.Drug groups were treated with DAMGO or morphine(10-12-10-4mol·L-1)for 15 min,then LI was detected.A:DAMGO reduced β-arrestin 2 recruitment in H6.52 and Y7.43 mutant MOR;B:morphine did not change β-arrestin 2 recruitment in H6.52 and Y7.43 mutant MOR compared with WT.β-Arrestin 2 recruiment=LI of drug group-LI of blank group.±s,n=3.
3 讨论
本研究结果表明,Y7.43突变后,相较于野生型MOR,DAMGO和吗啡对β-制动蛋白2的招募消失。2014年Hothersall团队从MOR激动剂与受体结合的机制着手,首次研究了MOR的关键氨基酸残基Y7.43在影响信号通路中的作用,结果证实吗啡不能引起WT和Y7.43F突变型MOR下游β-制动蛋白依赖性信号通路的激活[10],这与本研究得到的结论一致,提示Y7.43位点对于稳定β-制动蛋白2偶联的活性受体构象具有重要作用。该结论在胆囊收缩素2受体的研究结果中同样得到证实[11]。
另外,在氨基酸位点突变时,本研究考察了氨基酸的酸碱性、亲水亲脂性和位阻3个因素在影响药物与MOR结合中可能发挥的重要作用。例如,将H6.52突变为L,D和W 3种氨基酸,是将组氨酸的咪唑侧链替换为直链氨基酸、酸性氨基酸和具有增大位阻功能的吲哚基团;将Y7.43突变为A,F和S 3种氨基酸,是将酪氨酸的R基替换为甲基、苯环和羟甲基。
在化合物与MOR结合层面,本研究结果表明,H6.52突变使得DAMGO和吗啡均不能与MOR结合,而Y7.43突变后不改变DAMGO和吗啡与MOR的结合,表明H6.52是影响DAMGO和吗啡与MOR结合的位点。综合配体、受体结合后产生的效应和配体、受体分子对接结果分析得出,DAMGO与MOR分子对接时,DAMGO苯环上的酚羟基与D3.32 R基的羰基氧形成氢键,Y7.43的酚羟基与D3.32的羰基氧形成氢键,即形成3-7锁;吗啡与MOR分子对接时,吗啡结构中苯环上的羟基通过水桥同时与H6.52的咪唑侧链和K5.39的羧基氧相互作用,因此存在较复杂的氢键网络,此外,Y7.43的酚羟基也与D3.32的羰基氧形成氢键,体系中存在3-7锁。信号通路活性测定实验结果表明,DAMGO和吗啡均减弱下游G蛋白和β-制动蛋白通路的激活程度,推测H6.52位点通过影响DAMGO和吗啡与MOR的结合来发挥作用,同时氢键网络也可能参与其中;相对于H6.52D和H6.52W,H6.52L几乎阻断了DAMGO和吗啡对下游G蛋白依赖性信号通路的激活,推测H6.52位点突变为亮氨酸阻断了原组氨酸的咪唑环与配体小分子的氢键作用。当Y7.43位点突变后,使得体系中不存在3-7锁,细胞实验结果表明,Y7.43位点突变后,较野生型相比,DAMGO和吗啡减弱MOR下游G蛋白和β-制动蛋白通路的激活效应,由此推测3-7锁是影响G蛋白和β-制动蛋白依赖性信号通路激活的关键因素,而对于吗啡不激活野生型MOR下游β-制动蛋白依赖性信号通路,推测可能存在其他的相互作用参与此信号通路的激活,具体的作用机制还需要进一步的分子动力学模拟证实。
综上所述,Y7.43是造成DAMGO和吗啡对MOR下游信号通路差异性变化的位点,说明位点引起信号通路的变化不一定是位点干扰配体与受体的结合,而是不同的配体与受体具有不同的结合模式,即受体构象学差异,因此,残基在形成活性受体构象中具有重要的作用,残基突变则可以定性改变与配体结合后的受体激活方式。H6.52和Y7.43在一定程度上解释了配体如何激活受体以及受体结合口袋在配体与效应分子进行耦联过程中所发挥的作用。
猜你喜欢
介入放射学杂志(2022年11期)2022-03-04
江西农业学报(2021年4期)2021-04-20
医药前沿(2020年23期)2020-12-03
中成药(2018年10期)2018-10-26
材料科学与工程学报(2016年4期)2017-01-15
合成化学(2015年4期)2016-01-17
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
西南军医(2015年6期)2015-01-23
西南军医(2015年2期)2015-01-22
