
胱氨酸病婴儿型(肾病型)1 例报告并文献复习
2021-12-27 04:38:54崔洁媛葛兰兰张东风韩佩桐袁晓颖
临床儿科杂志 2021年12期
崔洁媛 葛兰兰 张东风 刘 玲 韩佩桐 袁晓颖
河北省儿童医院肾脏免疫科(河北石家庄 050031)
胱氨酸病(cystinosis)是一种罕见的常染色体隐性遗传的全身系统性疾病,因编码L-胱氨酸转运蛋白(cystinosin)的CTNS基因(17 p 13.2)变异所致。L-胱氨酸转运蛋白是一类溶酶体胱氨酸-质子共同转运体,其缺乏导致溶酶体无法将胱氨酸正常转运出去,从而导致胱氨酸在溶酶体中累积并逐渐形成结晶[1]。根据基因变异致病性、发病年龄以及肾脏受累的严重程度,胱氨酸病可分为三种类型:婴儿型(肾病型,OMIM #219800)、青少年型(中间型或迟发型,OMIM #219900)及成人型(眼病型,OMIM #219750)。临床以婴儿型(肾病型)最为常见,约占95%,临床主要表现为范可尼综合征(Fanconi syndrome)、慢性肾功能不全至终末期肾脏病(end stage renal disease,ESRD)[2]。本文报告1 例婴儿型胱氨酸贮积症,并结合相关文献进行 分析。
1 临床资料
患儿,男,2岁,因发现身高增长缓慢1年余,双下肢无力3个月入住河北省儿童医院肾脏免疫科。患儿系G2P2,足月顺产,出生体质量3.6 kg,身长50 cm,羊水、胎盘未见异常。生后混合喂养,按时添加辅食。3个月抬头,6个月独坐,8个月会爬,1岁会走。1岁时家长发现患儿身高较同龄儿偏矮,1 岁6 个月时身高76.7 cm、体质量9.3 kg。1岁9个月时出现双下肢无力,行走困难、步态不稳,呈鸭步步态,无发热,无多饮、多尿,无呕吐、腹泻,无手足搐搦,智力发育与同龄儿相仿。家长自行间断口服水解蛋白和维生素D无明显改善。
入院查体:P 95次/min,R 24次/min,身高78.7 cm(<P3)、体质量10 kg(<P3)。神清,精神反应好,部分牙齿牙釉质发育不良。胸廓两侧对称,肋缘外翻,胸骨中下段凹陷。双腕关节钝圆形环状隆起似手镯。心肺腹及神经系统未见异常。
辅助检查:X 线片示左腕骨骨龄相当于1 岁,尺桡骨远端稍膨大,双侧股骨远端干骺端增宽,双侧胫腓骨骨密度较低,双侧胫腓骨近端及远端干骺端稍宽;尿常规示pH值6.0,葡萄糖(++++),蛋白(++),酮体(++);血生化示血磷0.62 mmol/L,碱性磷酸酶 5 217 U/L,二氧化碳12.1 mmol/L,氯110.8 mmol/L,钠135 mmol/L,钾3.72 mmol/L,离子钙1.07 mmol/L;血气分析示pH 值7.20;24 小时尿蛋白0.27 g(27 mg/kg),24小时尿钙2.40 mmol(0.24 mmol/kg);肾早期损伤指标示尿转铁蛋白6.29 mg/L,微量白蛋白 141 mg/L,α1微球蛋白157 mg/L,N-乙酰β-D氨基葡萄糖苷酶(NAG酶)23.65 IU/L;尿蛋白电泳示白蛋白45%,小分子蛋白55%;血代谢筛查示游离肉碱缺乏,尿代谢筛查提示酮尿(乳酸、2-羟基丁酸、丙酮酸及3-羟基丁酸增高);尿氨基酸分析示多种氨基酸水平升高;骨密度示Z 值-2.1 SD;血25-羟基维生素D3正常低限(25.7 ng/mL)。血常规、心肌酶、肝肾功能、血糖、自身抗体、免疫球蛋白、补体、甲状腺功能、甲状旁腺功能、血氨、乳酸、铜蓝蛋白、胸片、心电图、彩色多普勒超声心动图、肾脏超声、头颅MRI 等未见异常。眼科会诊角膜未见异常。Gesell DQ评分91分,提示神经运动发育评估基本正常。
根据患儿临床表现初步诊断为范可尼综合征[3]。给予磷酸盐合剂、枸橼酸钾口服液(钾离子2 mmol/kg)、阿法骨化三醇、左卡尼汀等对症治疗1 周,患儿酸中毒纠正(二氧化碳24.6 mmol/L),血磷升至0.98 mmol/L,碱性磷酸酶降至424 U/L。因患儿发病年龄早,原发病方面考虑遗传代谢性疾病。患儿家属签署知情同意书后,取患儿及父母静脉血样本,行二代测序(遗传性肾脏疾病基因包)基因变异分析结果示患儿CTNS基因复合杂合变异,一个为剪接变异c.140+1 G>C(父源),另一为错义变异c.969 C>G(p.N323K,母源),Sanger测序验证结果见图1、2。明确诊断为胱氨酸病(婴儿型)。
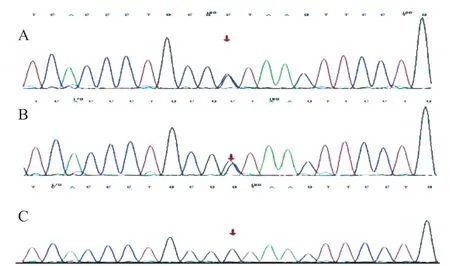
图1 CTNS 基因变异(c.140+1G>C 杂合变异)
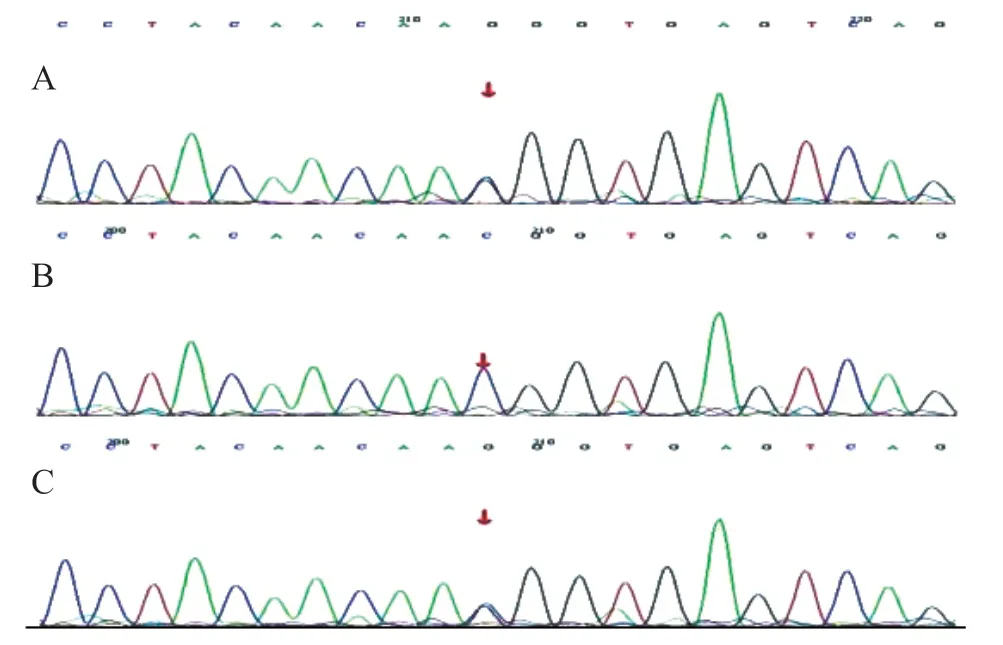
图2 CTNS 基因变异(c.969C>G 杂合变异)
2 讨论
胱氨酸病为一种罕见的常染色体隐形遗传性溶酶体贮积病,特征为胱氨酸在不同器官和组织中蓄积,导致可能很严重的器官功能障碍,因此临床表现为多系统、器官受累,包括肾脏、角膜、骨胳、甲状腺等。国外报道胱氨酸病的发病率约为1/20万~1/10万,国内仅有个例报道[4-6]。
通过PubMed(https://pubmed.ncbi.nlm.nih.gov/)、万方医学网(https://www.wanfangdata.com.cn/)及中国知网(https://www.cnki.net/)等进行文献检索,并结合国内外文献进行复习。
临床表现方面,因为发现或就诊的年龄不同,所以首发临床表现各异,但总体来说,年龄小者以体格发育落后和尿检异常为主[4-5]。国内文献报道明确基因诊断者12 例,其中6 例(50%)为体格发育落后;3例(25%)尿检异常,糖尿和/或蛋白尿;其他胃肠道表现(呕吐)、肢体无力和角膜异常各1 例(8.13%),但该3例发病时尿常规均异常,即糖尿和/或蛋白尿。国内报道的12 例患儿主要临床表现包括体格发育落后11/12(91.67%)、肢体无力5/6(83.33%)、佝偻病11/12(91.67%)、代谢性酸中毒12/12(100.00%)、低钾血症5/6(83.33%)、低磷血症4/6(66.67%)、角膜病变3/6(50.00%)、多饮/多尿4/6(66.67%)和糖尿/酮尿/氨基酸尿/蛋白尿12/12(100.00%),肾结石1/12(8.33%)和肾功能异常2/12(16.67%)相对少见,可能与随访年限较少、年龄相对较小有关。本例患儿,首发临床表现也是体格发育落后,此外其他主要临床表现包括佝偻病、代谢性酸中毒、低钾血症、低磷血症和糖尿/酮尿/氨基酸尿/蛋白尿,均与文献结果相符[7-9]。
临床分型方面,国内文献报道的12例患儿,婴儿型10/12(83.33%),青少年型2/12(16.67%)。本例患儿也是婴儿型提示胱氨酸病临床分型以婴儿型最为常见,可高达95%。需要注意的是,由于胱氨酸病临床表现缺乏特异性且累及多系统,早期主要症状为尿检异常如糖尿、氨基酸尿等,不容易发现,因此临床诊断较为困难,诊断年龄均偏晚,不能单纯以发现临床症状或诊断时年龄作为临床分型的依据,应结合患儿累及器官、系统及病史进展、预后综合考虑。
基因型和临床表型方面,关于胱氨酸病基因型和临床表型相关性的研究显示,同时发生于两个等位基因的截短变异或对蛋白功能影响较大的变异类型,患者往往表现为婴儿型胱氨酸病且病情较重,而携带轻微变异类型者则表现为青少年型或成人型[10-13]。该观点有待于大样本、多中心研究进一步证实,如c.969C>G(p.323N>K)纯合变异,国内多数研究报道包括本病例均为婴儿型胱氨酸病,但国外报道中也有临床表型为青少年型胱氨酸病者,提示相同变异可以引起不同的临床表型。其原因一方面可能与人种差异有关,另一方面也可能与临床表现隐匿、确切发病年龄难以确定、诊断时间偏差及分型标准欠准确等有关。此外,同一家系基因型完全相同的患儿,临床表型也可有差异[14]。
猜你喜欢
成都医学院学报(2021年2期)2021-07-19 08:35:32
环境污染与防治(2021年11期)2021-01-09 02:08:51
临床儿科杂志(2020年2期)2020-03-12 04:04:10
发明与创新(2019年42期)2019-11-18 01:24:04
华声文萃(2019年8期)2019-09-10 07:22:44
文萃报·周二版(2019年25期)2019-09-10 07:22:44
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国医疗美容(2015年4期)2015-04-27 02:24:11
