
以头痛为主要表现的Ⅰ型神经纤维瘤病1 例报告
2021-12-27 04:38:56汤继宏张兵兵师晓燕邵艺华
临床儿科杂志 2021年12期
徐 欢 汤继宏 肖 潇 张兵兵 师晓燕 邵艺华
苏州大学附属儿童医院神经内科(江苏苏州 215025)
I型神经纤维瘤病(neurofibromatosis type 1,NF1)是一种常染色体显性遗传的皮肤神经综合征,是神经纤维瘤病中最常见的一种,儿童患病率约为1/3000[1]。NF1是由染色体17q11.2上的抑癌基因NF1变异引起,其蛋白产物,神经纤维蛋白在调控细胞生长和分化中起重要作用。NF1典型的临床表现包括皮肤牛奶咖啡斑、腋窝或腹股沟雀斑、多发神经纤维瘤、特征性骨性病变及视神经胶质瘤等。多数NF1患儿可依靠临床表现及阳性家族史得到确诊,但部分患儿症状不典型或处于早期,需结合影像学和基因检测等手段协助诊断。本文总结分析1例以反复头痛为主要表现的NF1患儿的临床资料,并结合文献复习进行讨论,以期对NF1的多样性及诊治提供一定借鉴作用。
1 临床资料
患儿,男,10岁1个月,因反复头痛1年入院。患儿入院前1 年无明显诱因下出现反复头痛,主要集中在双侧顶部,性质模糊,为轻中度(疼痛视觉模拟评分法2~4 分,10 分制),持续数秒至数分钟不等,1~4 次/ 月,紧张或活动可加重,休息后能稍缓解,不伴恶心呕吐、畏光畏声、头晕耳鸣,与体位无明显关系。多次至当地医院就诊,考虑血管神经性头痛,予对症处理后效果不佳。近1 年来患儿述头痛发作较前频繁,1~3次/周,性质同前,期间查头颅CT未见异常。为进一步诊治来苏州大学附属儿童医院神经内科住院。患儿既往史无特殊,为G1P1,足月顺产,出生体质量3 400 g。患儿运动、语言及生长发育与同龄儿一样。患儿父亲及奶奶躯干亦有散在褐色斑块及多发隆起结节,但均未就诊,也无特殊不适,生活及工作未受影响。
体格检查:体温36.0 ℃,脉搏98 次/min,呼吸18次/min,体质量32 kg,身高 142 cm,收缩压/舒张压(SDP/BDP)106/70 mmHg;神志清,精神可,反应灵敏,定向力、记忆力、计算能力等无异常。患儿近1~2 年出现右眉弓上、左锁骨上及龟头隆起结节,渐进性增大,各个隆起结节质地硬,边界欠清,活动度稍差,有轻压痛,表面皮肤无红肿,未及波动感,直径约 5~10 mm;前胸及后背可见近10枚散在褐色斑块,最大直径约8 mm,1 岁左右出现。心肺腹检查无特殊。四肢肌力及肌张力无异常,未见皮肤感觉异常,病理征未引出,脑膜刺激征阴性。
实验室检查:血常规、血生化、肿瘤标志物、血及尿遗传代谢产物检测、铜蓝蛋白、脑脊液常规及生化均无异常。眉弓B 超示右眉弓皮下探及大小约 11 mm×6 mm×3 mm 无回声团,边界清,透声可,后壁回声增强;彩色多普勒超声未探及血流信号,即右眉弓囊性包块。头颅MRI 平扫示两侧基底节区异常信号,两侧丘脑形态稍饱满伴信号异常(等T 1 长T2、Flair序列高信号影,图1)。脊柱MRI平扫示左侧C2-C3椎间孔及两侧腋下臂丛神经走形区异常信号(图2)。

图1 头颅MRI 平扫表现
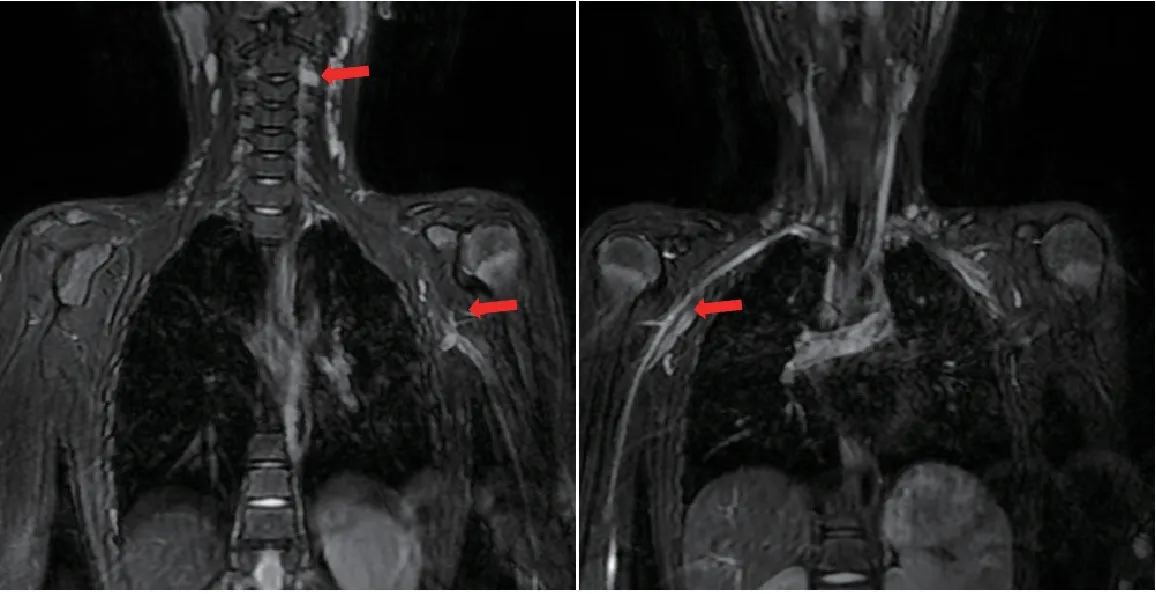
图2 全脊柱MRI 表现
结合患儿临床表现、家族史及影像学表现,考虑NF 1 可能。但患儿头痛病程较长,治疗效果不佳,经医院医学伦理审核,家属签署知情同意书后行全外显子家系检测。结果显示,患儿NF 1基因外显子c.6177_6183del TTTAGCA(p.Leu2060Alafs*4)变异,此变异以往未见报道,亦未在 HGMD 与 gnomAD等数据库中收录,为新发现的杂合变异。其父亲存在相同杂合变异,母亲未检测到变异基因(图3)。

图3 患儿及父母一代测序验证图
2 讨论
NF 1是由位于17 q 11.2 号染色体上的NF 1基因杂合变异引起,该基因共包含60个外显子,编码一种叫神经纤维蛋白的抑癌蛋白。该蛋白在多种细胞中表达,主要存在于神经元、施旺细胞及胶质细胞,在控制细胞增殖、分化、凋亡和迁移中发挥重要作用[2]。其中,约90%~95%的NF 1变异是基因内变异,少数是包含整个NF1基因(whole-gene deletion,WGD)及其侧翼序列的大片段缺失[3-4]。目前WGD被定义为以下4种:第一种缺失长1.4 Mb,包含14个基因,通常是遗传性变异;第二种缺失长1.2 Mb,通常发生于受精卵形成之后;第三种长1.0 Mb,包含9 个基因,是非常罕见的一种变异;第四种,把缺失的基因长度及涉及基因都有巨大异质性的一类归为不典型缺失,它的变异起源不固定。这四种WGD 定义可以帮助临床了解遗传性变异类型,也有助于病情判断及预后分析。多数NF 1基因内变异表现为片段截短,包括移码变异、无义变异及剪接变异,少数可表现为基因内某段拷贝数的变异、插入变异或错义变异。该患儿的基因报告显示其NF1基因片段内部缺失了一段TTTAGCA,为杂合变异,导致氨基酸序列2060 位由亮氨酸变为丙氨酸,且随后移码并在3 个氨基酸后终止翻译,可影响神经纤维蛋白的表达。该变异为新发现的变异(未在 HGMD 与 gnomAD 等数据库中收录,gnomAD:ALL:0.000%),按照ACMG 变异分类标准,可以归类为“可能致病性”变异。结合患儿临床表型及家系成员情况可明确NF1诊断,患儿该疾病来源于其父亲的致病变异。
虽然NF1是一种常染色体显性遗传病,但临床上有多达50%的NF 1 患者没有任何家族史,而是通过自发变异获得[5]。目前,人类基因变异数据库中存在2 600多个不同的NF1基因变异点。不同的变异具有相当大的异质性,甚至在那些携带相同家族变异的亲属中临床表现也有很大差异,这可能与NF1基因修饰及肿瘤组织继发体细胞变异有关。由于NF 1基因庞大且临床表现异质性极大,目前仅有较少的基因-表型关系建立。如上面第一种WGD 常表现为患儿过度生长、认知功能发育严重落后、先天性心脏病,及相较于其他NF 1 患儿显著增多的神经纤维瘤。并且这部分患儿发生恶性周围神经鞘瘤的风险也明显增高。NF1基因17号外显子c.2970-2972delAAT变异的患儿,常表现为单纯的皮肤牛奶咖啡斑和皮肤褶皱处雀斑,很少出现神经纤维瘤[6]。NF1基因29号外显子的1809 号密码子发生错义变异常会导致多发的皮肤牛奶咖啡斑、视神经通路胶质瘤、身材矮小、学习能力障碍、肺动脉瓣狭窄及努南综合征的表现(特殊遗传病面容、身材矮小、先天性心脏病、血液及骨骼系统并发症)。这类患儿可有或无皮肤褶皱处雀斑及虹膜Lisch结节,一般没有明显的神经纤维瘤[7-8]。此外,NF1基因844-848号密码子的错义变异一般与浅层丛状神经纤维瘤、脊柱神经纤维瘤、视神经通路胶质瘤、骨骼病变及更高风险的恶性肿瘤相关[9]。最近,有学者又报道了NF1基因p.Arg1038Gly错义变异与一种特殊皮肤表型相关,而不涉及神经纤维瘤或其他并发症[10]。在Legius综合征中发现的SPRED1基因杂合变异也可导致与NF1疾病类似的临床表现,该基因编码另一种抑癌蛋白,但该病的临床表现相对较轻,需依靠基因检测相鉴别。NF 1 具有相当大的表型异质性,提示临床医师要仔细查体(尤其是皮肤)、及时行神经影像学检查和基因检测。本患儿早期因反复头痛于当地医院就诊,体格检查未发现皮肤牛奶咖啡斑,头颅CT正常,临床医师即排除了颅内器质性病变。患儿亦曾因“龟头隆起结节”就诊当地泌尿外科,未予明确诊治方案。多次就诊经历不仅延误了其诊疗,也增加了患儿家庭的经济和心理负担。
NF 1 患者患各种良恶性肿瘤的风险要远高于正常人[11]。通常,影响NF1患者生活质量的主要是丛状神经纤维瘤,它由多种类型细胞组成,包括神经轴突、施旺细胞、成纤维细胞、肥大细胞、巨噬细胞、血管及细胞外基质[12]。约50%的NF1患者存在该类型肿瘤,可累及脊髓神经根到末梢神经的任何部位,最常见部位有脊柱旁(31%)、头颈(31%)和四肢(25%)[13]。这些肿瘤虽然是良性,但是常会引起神经痛、感觉减退、肢体活动障碍等问题。NF 1 出现恶性外周神经鞘瘤的发生率为10%,通常在40岁前出现,往往成为这部分患者的最终死亡原因[14-15]。除视神经胶质瘤外,NF 1 患者颅内病变主要累及基底节区、脑干及小脑,可引起头痛、头晕、恶心、癫痫、共济失调、肌张力障碍或局灶神经功能缺损症状[16]。本病例有较多结节,累及右眉弓上、左锁骨上及龟头等表皮部位,还累及了脊柱椎间孔及两侧腋下臂丛神经,此外,累及基底节区的颅内病变是患儿反复头痛的主要原因。远期随访要注意患儿这种多发异常组织增生及其造成的相应临床表现。NF 1 患者还可合并特征性的骨骼畸形、认知行为缺陷、心血管异常等问题[17-19]。NF1相关的骨科疾病多出现在儿童早期,表现为脊柱侧弯、骨质疏松、胫骨发育不良、蝶骨发育不良等。NF 1 患儿较低的智力水平、注意力困难、多动症和自闭症谱系障碍等问题可一定程度上影响这类人群的社会参与能力。虽然本病例目前仅有“反复轻或中度头痛”表现,尚未出现影响其生活、学习、运动和语言功能情况,正如其父亲及奶奶那样无特殊不适,未影响寿命和社会生活等,但仍然要多方面关注其将来病情发展情况。由于目前NF 1 尚无较好的治疗方案,嘱该患儿定期随访。
NF1患者管理内容涉及广泛,需定期检测患者的神经系统、心血管系统、骨骼系统及皮肤等,如出现不明原因腹痛还需警惕胃肠道肿瘤。这类患儿还可能存在原发性高血压、自闭症谱系疾病等众多问题。一般如果检查发现异常却没有明显临床症状,可不考虑手术切除病变。因为肿瘤血管丰富,切除有风险,且其复发概率较高。但是如果包块有明显占位效应,或随访发现有快速增长,或有持续性的疼痛,要警惕恶变的可能。2020年4月10日,美国FDA批准selumetinib (商品名:Koselugo)用于治疗小儿相关的有症状但无法手术的丛状神经纤维瘤。该药是强效的肿瘤信号通路相关酶抑制剂,发挥抑制肿瘤细胞生长的效果[20]。目前该药上市时间仍较短,临床效果及不良反应还需更多反馈资料论证。
猜你喜欢
现代装饰(2022年3期)2022-07-05 05:56:36
中老年保健(2021年10期)2021-11-30 09:34:06
皮肤病与性病(2021年3期)2021-07-30 08:07:40
基层中医药(2021年4期)2021-07-22 07:15:28
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
特别健康(2018年3期)2018-07-04 00:40:10
红土地(2016年11期)2017-01-15 13:46:38
少儿科学周刊·儿童版(2016年2期)2016-03-19 11:54:12
百科知识(2015年18期)2015-09-10 07:22:44
