
ITGA7 基因变异致先天性肌病1 例报告并文献复习
2021-12-27 04:38:58王春霞段丽芬王惠萍王左华
临床儿科杂志 2021年12期
孙 莹 王春霞 李 磊 段丽芬 王惠萍 王左华 张 霞
昆明市儿童医院1.神经内科,2.神经电生理中心(云南昆明 650000)
先天性肌病(congenital myopathies)是一组由基因变异所导致的病程相对静止的肌肉病,临床表现为不同程度的肌无力、肌张力低下、运动里程碑延迟。此外,脊柱侧凸、漏斗胸、髋关节脱位或畸形足也是先天性肌病常见临床表现,患者大多智力正常,中枢神经系统通常不受影响,血清中的肌酸激酶水平轻微升高或正常,肌电图大多显示肌源性改变,但也可正 常[1]。虽然很多先天性肌病已经明确了基因诊断,但更多潜在的致病基因仍有待被发现,现回顾性分析1例ITGA7基因变异致先天性肌病患儿的临床资料,并结合相关文献及已报道病例进行讨论。
1 临床资料
患儿,男,10 岁9 个月。因下蹲困难5 年余,于昆明市儿童医院神经专科门诊就诊。患儿5年余前在无明显诱因下出现下蹲困难,表现为下蹲动作时小腿肌肉及跟腱处强烈的紧缩感,强行下蹲时脚后跟不能着地,影响如厕,但不影响日常活动,可正常行走、跑步、爬楼、上体育课,病情无进行性加重,认知语言正常,学习成绩优异。神经系统查体:神清,对答切题,定向力、计算力正常,颅神经检查无异常,行走时左脚轻微内收,下蹲时左右脚间距增大,脚后跟不能着地,跟腱挛缩,踝关节背屈轻度受限,腓肠肌无肥大,肌肉容积正常,Gower征阴性,四肢肌张力正常,肌力正常,四肢膝反射正常,巴氏征(-),脑膜刺激征阴性。患儿为G2P2,足月顺产,无围生期缺氧窒息、脑损伤病史,自幼生长发育正常,父母体健,非近亲,无遗传病家族史。实验室检查:血常规、肝肾心功能、电解质、甲状腺功能、乳酸、铜蓝蛋白,血浆氨无异常,血清肌酸肌酶(creatine kinase,CK)升高286 U/L。头颅、腰骶椎磁共振成像(MRI)无异常。检测下肢股神经、胫神经、腓总神经的运动传导速度、潜伏期、复合肌肉动作电位波幅、F波,结果均未见异常。针极肌电图检查双层胫前肌、腓肠肌、股四头肌肌电,发现轻收缩双侧胫前肌MUAP波幅降低,时限缩短,多相电位显著增多,大力收缩呈病例性干扰相,呈肌源性损害 表现。
患儿起病缓慢、病程长,临床表现不典型,以下蹲时姿势异常及血清CK 轻度增高为主要表现,肌电图提示肌源性损害表现,考虑肌病可能,家长拒绝完善肌肉活检,故为进一步明确诊断,经患儿家属知情同意,用乙二胺四乙酸(EDTA)抗凝管抽取患儿及父母外周静脉血各3 mL,混匀后置-80 ℃冰箱保存送北京康旭医学检验所有限公司进行医学全外显子检测行医学全外显子检测。发现患儿的ITGA7基因存在c.1100dupG纯合移码变异,受检者其父母该位点均为杂合子,检测结果经Sanger 测序验证(图1)。该变异不属于多态性变化,在人群中发生的频率极低(所参考数据库:1000Genomes、dbSNP),美国医学遗传学与基因组学会(ACMG)评分为PVS1、PM2、PP5,为致病性变异,该变异尚未见文献报道。结合临床表现,患儿诊断为 ITGA 7 缺乏相关先天性肌病,遗传方式为常染色体隐性遗传。
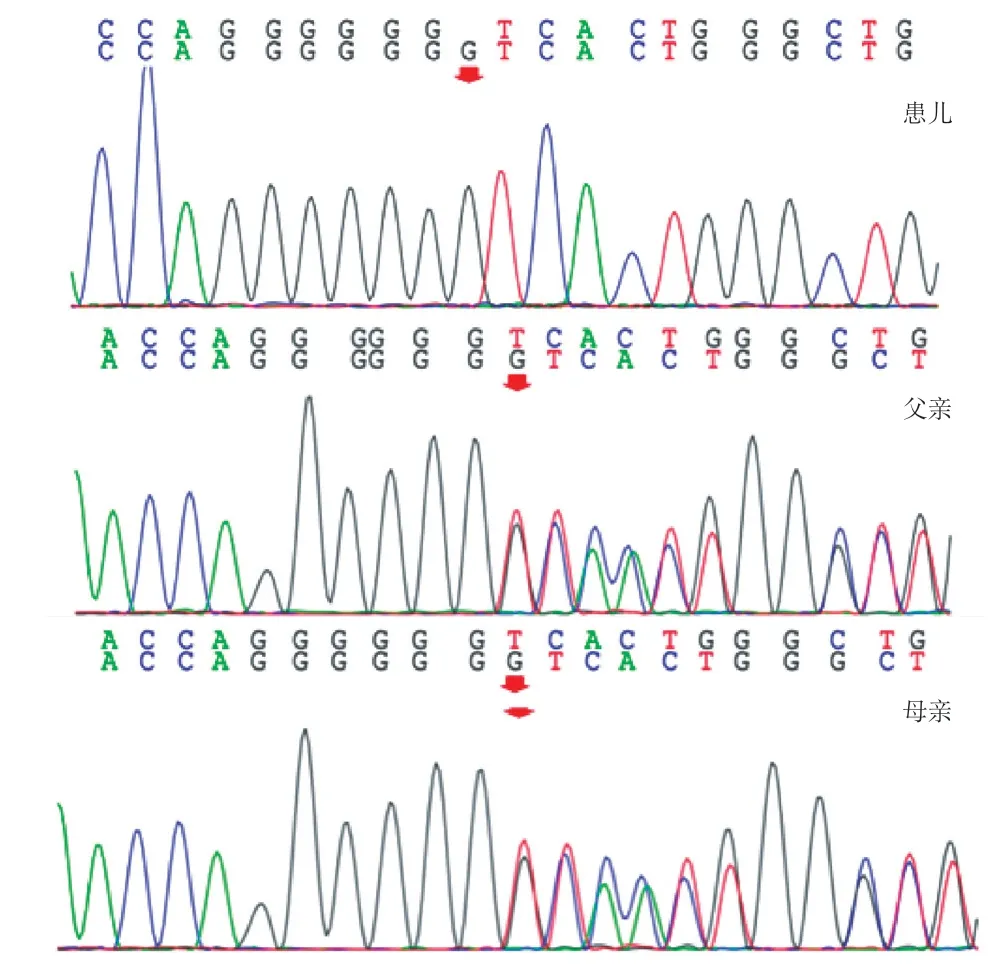
图1 ITGA7 基因Sanger 测序图
2 讨论
整合素是一组异二聚体膜糖蛋白受体,介导细胞外基质和细胞骨架蛋白间的联系[2]。整合素α 7 β 1就是其中一种表达于骨骼肌的跨膜层粘连蛋白受体,与层粘连蛋白α2链(laminin alpha-2 chain,LAMA2)及抗肌萎缩蛋白-糖蛋白复合物结合,提供肌肉纤维和基底膜之间的机械连接,发挥各种功能,如肌细胞的迁移和增殖、神经肌肉和肌腱连接的形成,以及对产生收缩力至关重要的肌肉纤维的“黏合”等。整合素α7β1功能的改变与多种肌病相关[2]。
ITGA7主要在肌腱膜连接处表达,由ITGA7基因编码,ITGA7基因变异可引起ITGA7缺乏,导致先天性肌病[3]。
1995年,人类ITGA7基因被发现并定位于染色体12 q 13[4]。ITGA 7基因由至少27 个外显子组成,跨越约22.5 kb的区域。1997年,Mayer等[3]在小鼠种系中产生了ITGA7基因的一个空等位基因,变异纯合子小鼠出生后不久出现肌无力表现,超微结构上,肌纤维的肌腱连接失去交错,肌丝从肌膜上缩回,肌腱连接功能受损,证实了ITGA7基因变异可引起肌纤维结构及功能的破坏。
已有大量研究证实ITGA 7在其他常见肌营养不良发生发展中的作用。LAMA 2基因变异致 LAMA 2 缺乏是临床最常见的先天性肌营养不良,可引起严重肌张力减退、肌无力、关节挛缩、认知障碍等,在其患者及动物模型中发现,ITGA7表达也显著下降,推测ITGA 7 的功能障碍是导致LAMA 2 严重表型的基 础[5]。杜氏肌营养不良(Duchenne muscular dystrophy,DMD)是儿童期最常见的进行性肌营养不良,由DMD基因产物抗肌萎缩蛋白丢失所致,在DMD 患者和其大鼠模型中可以看到ITGA 7 介导的细胞外基质连接增强,一定程度上弥补了抗肌萎缩蛋白缺失导致的的肌肉完整性破坏,因此,ITGA7被认为是DMD症状的重要调节器[6]。有学者通过过度表达ITGA7来替代抗肌萎缩蛋白的缺失,以此来治疗DMD,被证明是一种治疗DMD的前景[7]。
虽然ITGA 7 的功能及其在常见肌营养不良发生发展中的作用已被熟知,但ITGA 7 缺陷本身引起的先天性肌病却非常罕见。截止2020 年12 月,共报道ITGA 7基因变异相关病例5 例[8-11],结合本例共6 例患者,共发现ITGA7基因6种不同变异类型,其中,内含子区剪切变异2个,外显子区移码变异2个、错义变异2 个(表1)。6 例患者中3 例患者病情相对稳定,均因运动发育落后或步态异常而就诊,另外3 例患者病情呈进展性,以肌无力、肌张力减低为主要表现,其中2 例生后不久起病,1 例因呼吸衰竭死亡,1 例合并左心室非压实性心肌病并因心源性因素死亡。此外,先天性髋关节脱位、斜颈、脊柱侧弯、关节挛缩也是其常见临床表现,ITGA7基因变异相关肌病患者认知功能多正常,所有患者CK均轻度增高(163~528 U/L),见表2。

表1 ITGA7基因变异相关病例的基因型
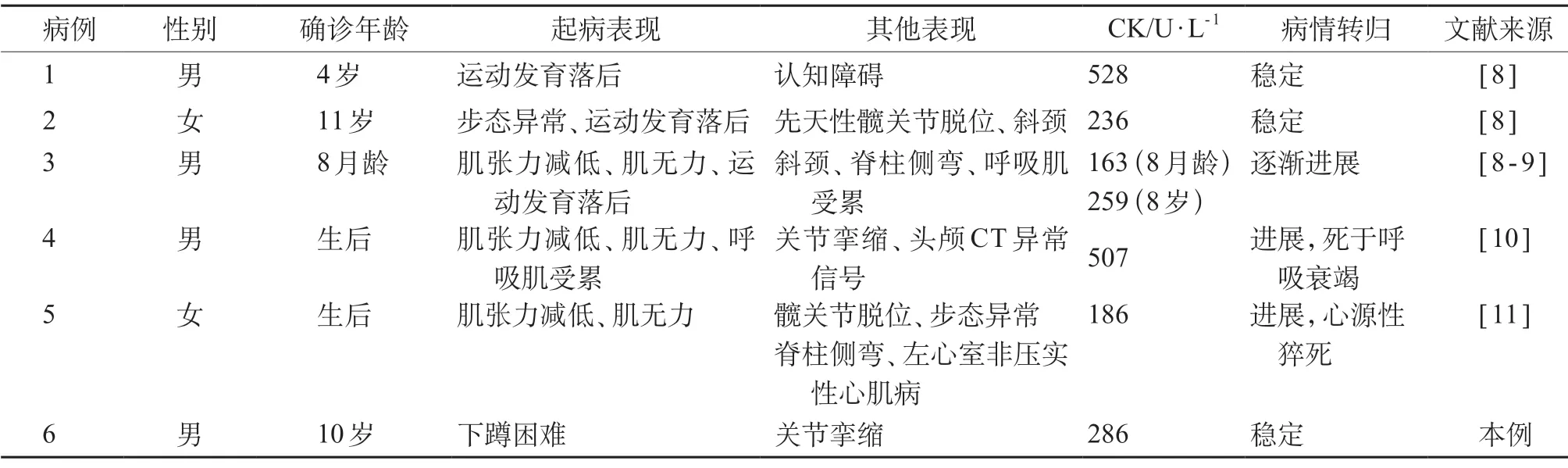
表2 ITGA7基因变异相关病例临床表型
1998年在117例原因不明的先天性肌病和先天性肌营养不良患者的肌肉活检中检测ITGA7的表达,首次发现3 例ITGA 7基因变异所致肌病患者[8],3 例患者肌肉ITGA7表达显著下降,肌肉病理可见轻度的肌肉纤维大小的变化,I型纤维占优势,很少或没有肌纤维的坏死和再生,与动物实验[3]的结果相似,故认为ITGA7基因相关疾病应该被称为先天性肌病而不是先天性肌营养不良(congenital muscular dystrophy)。先天性肌病病程相对静止,肌肉病理以肌纤维结构异常为主,很少见到肌肉坏死或再生[1],而先天性肌营养不良肌无力常为进展性,肌肉活检可见肌纤维坏死和再生、脂肪侵润等[12]。但随后对其中1例患者的长达10年的随访提示病情呈进展性[9],患儿肌无力逐渐加重,最终失去行走能力。另有报道1 例ITGA 7基因错义变异c.1969 C>G(Arg 657 Gly)患者,生后即出现呼吸肌无力,因呼吸衰竭死亡,肌肉活检可见肌纤维坏死,符合先天性肌营养不良表现[10]。故ITGA7基因变异相关疾病的分类及命名还有争议,本例患儿病情稳定,肌无力轻,以轻度跟腱挛缩及轻度肌酶升高为主要表现,应诊断为先天性肌病。
CK 异常升高常提示肌膜通透性的破坏,是临床辅助诊断肌肉病的敏感指标,先天性肌病患者CK 通常轻度升高或正常,而CK 升高5 倍以上需要高度警惕肌营养不良[12]。在已报道的ITGA 7 缺乏患者中,无论病情稳定或进展,肌酶均呈现正常或轻度增高(163~528 U/L),为其特征性表现。此外,肌酶轻度增高还可见于药物、炎症、运动、惊厥发作等,因此,诊断时还需要根据病史、其他临床表现做相应的鉴 别诊断。
2013年,意大利学者报告了一个家系[11],先证者发现了两个不同基因的纯合错义变异,即肌球蛋白重链7B (MYH7B)和ITGA7,该患者既有MYH7B基因变异相关左心室非压实性心肌病表现,又存在先天性肌病表现,股四头肌活检显示1型纤维占多数,其平均直径比2 型纤维小,这些发现与先天性纤维型不均衡(congenital fiber type disproportion,CFTD)表现一致。CFTD 是先天性肌病的一种,临床表现与许多其他先天性肌病相似:在儿童和青少年时期,肌肉无力通常相对稳定或缓慢发展,轻度跟腱挛缩多见。本文所报道病例肌无力表现不明显,进展缓慢,以下蹲困难为主要临床表现,且在下蹲时跟腱牵拉受限,推测与轻度跟腱挛缩相关,故本文所报道病例是否为CFTD表型尚需要进一步的肌肉活检确定。
和其他先天性肌病相似,对ITGA7基因变异相关肌病的治疗主要是针对肌无力所引起的并发症给予支持和对症治疗。
综上,ITGA 7相关肌病轻重不一,轻者为先天性肌病样表现,肌无力轻,进展缓慢,以运动发育落后、步态异常等为主要表现,重者呈先天性肌营养不良样表现,出生时即表现为严重肌张力低下、肌无力,病情进展快,但无论轻重,所有患者肌酶均仅为轻度增高,甚至正常。由于ITGA7基因变异相关肌病已报道病例数极少,还需要更多的研究及对患者的长期随访来阐述其临床特点及转归。本病例为国内首次报道的ITGA 7基因变异相关肌病,通过这个病例,让我们认识到了ITGA7在肌纤维结构及功能中的作用,以及其基因变异后致先天性肌病的临床表现,当临床上遇到CK值轻中度增高,表现为肌病的患者,需考虑到该病的可能,完善基因检查可帮助确诊。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
今日农业(2021年5期)2021-11-27 17:22:19
现代畜牧科技(2021年5期)2021-07-20 08:07:40
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:56
中国中医急症(2019年10期)2019-05-21 07:20:42
天津农学院学报(2016年2期)2016-12-01 05:40:05
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
