
以腹痛为主要表现的儿童自身炎症性疾病2 例报告并文献复习
2021-12-27 04:38:56赵雪奇闾佳佳王歆琼许春娣
临床儿科杂志 2021年12期
赵雪奇 闾佳佳 余 熠 王歆琼 许春娣 肖 园
上海交通大学医学院附属瑞金医院儿内科(上海 200025)
自身炎症性疾病(autoinflammatory diseases,AUIDs)[1-2],是在2000年左右被定义的一组疾病,因固有免疫失调而引起全身炎症反应,绝大部分属于单基因遗传病,主要包括家族性地中海热(familial Mediterranean fever,FMF)、A 20 单倍剂量不足(haploinsufficiency of A 20,HA 20)、肿瘤坏死因子(tumor necrosis factor,TNF)受体相关周期性综合征、冷炎素相关周期性综合征等。AUIDs以复发性全身性炎症为特征,临床症状主要包括周期性发热,反复发作的关节炎,口腔溃疡以及以腹痛、胸痛为特点的浆膜炎等。这些症状不仅常见,且均不具有特异性,一旦以单个症状为主要表现,则极易出现误诊误治。现报告2例以反复腹痛为主要表现的AUIDs患儿,以提高儿科医师对该病的认识。
1 临床资料
例1,男,9岁,汉族,G1P1,足月顺产,无窒息抢救史,出生体质量不详,出生后母乳喂养,否认家族遗传病史。现身高127 cm(P3~P10),体质量24 kg(P3~P10),体格检查未见明显异常。患儿6 岁6 月龄时因右下腹痛外院诊断为阑尾炎并行手术治疗。手术后患儿仍有反复腹痛,予对症处理症状缓解后出院。2个月后患儿再次因腹痛加重就诊于上海交通大学医学院附属瑞金医院。病程中患儿无反复发热、关节痛、呕吐腹泻、消瘦等表现;当时实验室检查:外周血白细胞(WBC)18.4×109/L,C反应蛋白(CRP)26.0 mg/L,病原学检查无阳性发现。后患儿多次腹痛时均出现外周血WBC、CRP 增高;TNF-α 明显增高,粪钙卫蛋白达1 250.0 μg/g;相关自身抗体包括抗核抗体(ANA)、抗中性粒细胞胞浆抗体(ANCA)、抗可提取核抗原(ENA)均阴性。胃肠镜检查未发现特征性变化。腹部CT及CT血管造影、小肠MRI均提示肠系膜淋巴结肿大(图1),同时排除消化道畸形、肿瘤和肝胆胰的相关疾病。予以抗感染治疗后患儿症状未见明显好转,考虑感染性疾病可能性不大,自身免疫性疾病无法排除。予甲基泼尼松龙25 mg/d 治疗,抗炎5 天后腹痛症状明显好转,复查血常规无异常后继续予以泼尼松 10 mg/d口服,患儿无腹痛发作与反复。服用泼尼松2周,家属自行停药后出现腹痛反复,但在腹痛时口服泼尼松5~10 mg/次即可缓解,以此方式治疗直至基因检测确诊之后停用,共6个月。
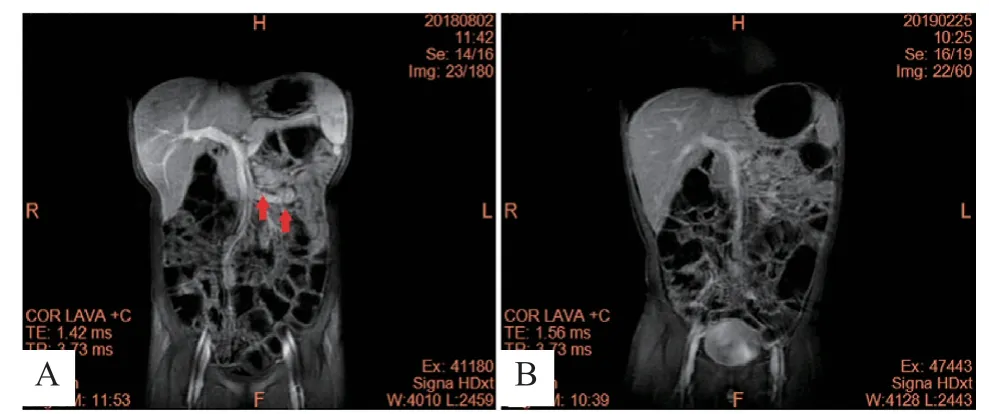
图1 例1 患儿腹部MRI 表现
例2,女,3 岁,汉族,G1P1,胎龄36+2周,剖宫产,无出生抢救史,出生体质量3.1 kg,生后母乳喂养;母亲幼年时有腹痛发作病史,长大后腹痛缓解。患儿身高92 cm(P3~P10),体质量12.5 kg(P3~P10),体格检查腹部稍膨隆,其余未见明显异常。患儿2岁9个月无明显诱因出现腹痛,脐上为主,每周发作3~4次,发作时较剧,转移注意力或解便后可缓解,与进食无明显关系,夜间腹痛,常于凌晨痛醒,数十分钟自行缓解。病程中无反复发热、口腔溃疡、关节疼痛、腹泻等表现。患儿腹痛发作时外院实验室检查示外周血WBC、CRP 明显增高。病原学检查无阳性发现,体格检查也未见明显异常,曾多次予以经验性抗感染治疗,仍有反复腹痛。本次入院实验室检查示外周血WBC 18.3×109/L、Hb 92.0g/L、血小板643.0×109/L、CRP 103.6 mg/L,白介素(IL)-1、IL-6及TNF-α均明显升高,粪钙卫蛋白水平>1 800.0μg/g,ANA、ENA、ANCA 等自身免疫性疾病相关抗体均为阴性。电子胃镜检查示出血性糜烂性胃炎、十二指肠多发性薄苔浅溃疡(图2)。结肠镜未见明显异常。十二指肠黏膜活检病理组织提示存在隐窝炎。小肠CT 示小肠节段性肠壁增厚,肠系膜淋巴结多发肿大(图3)。结合患儿病史考虑自身免疫性肠病可能,予泼尼松 5 mg/d治疗10天后腹痛仍有发作。
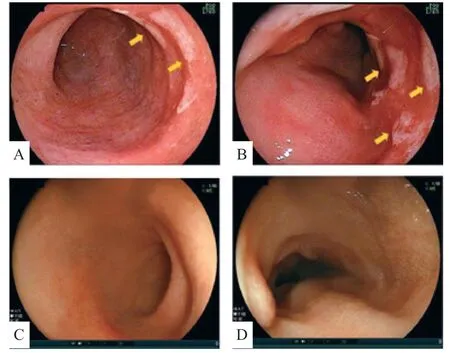
图2 例2 患儿胃镜表现
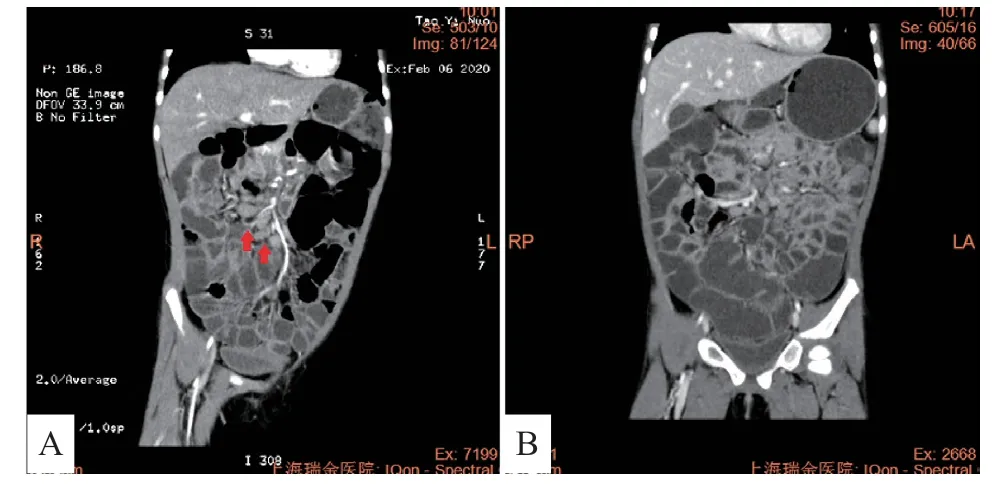
图3 例2 患儿腹部CT 检查结果
鉴于2 例患儿均以反复腹痛起病,发作时急性期蛋白和多种促炎细胞因子增高,疑似自身炎症性疾病。由于患儿的临床症状、实验室检查无特异性,故没有选择相关的靶基因二代测序,而直接进行了全外显子二代测序。在患儿监护人知情同意后,留取了患儿及其父母EDTA 抗凝血各2 mL,进行全外显子测序。结果例1 患儿TNFAIP 3基因存在杂合缺失变异(c.440-441del,p.Leu147Glnfs*7),导致TNFAIP3基因第440~441位核苷酸位点缺失,最终引起A20蛋白第147 位由亮氨酸变异为谷氨酸,并在继续翻译7 个氨基酸后肽链终止;变异未在患儿父母中发现,为新生变异(图4)。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)2015年的指南[3],该位点被评为致病性变异(PVS+PS2+PM2)。例1患儿诊断更正为TNFAIP3变异所致HA 20。例2 患儿MEFV基因第2 外显子存在杂合变异(c.726C>G,p.S242R),导致MEFV基因第726位核苷酸由胞嘧啶突变为鸟嘌呤,编码的242位氨基酸由丝氨酸变异为精氨酸;变异来自母亲。已有功能试验明确该变异可导致MEFV基因编码的蛋白质功能受损[4](图5)。该变异在人类外显子数据库、千人基因组和人群基因组变异频率数据库中均为低频变异。因此,根据2015年ACMG发布的指南[3],该位点被评为致病性变异(PS 3+PM 2+PP 1_Strong+PP 4)。例2患儿最终诊断为FMF。
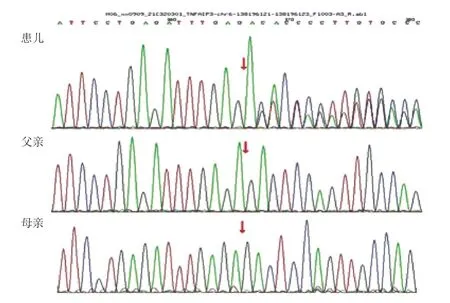
图4 例1 患儿及父母全外显子组高通量测序结果
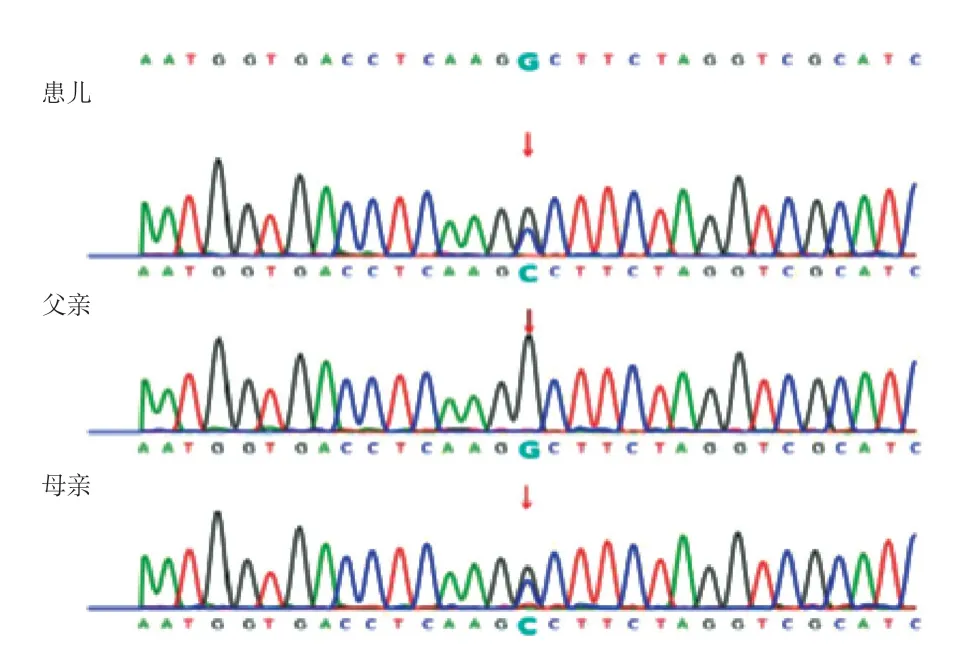
图5 例2 患儿及父母全外显子组高通量测序结果
例1 患儿自口服糖皮质激素治疗后,生长速率明显减慢,为4 cm/a,考虑为长期口服激素引起的生长缓慢,故诊断明确后改用秋水仙碱0.5 mg/d 口服,同时停用激素,患儿腹痛发作频率明显减少,血细胞因子和粪钙卫蛋白水平明显下降。生长速率恢复至 6 cm/a。例2 患儿逐渐减停泼尼松,给予秋水仙碱 0.25 mg/次,每天1次,即0.02 mg/(kg·d)。用药3日后患儿腹痛发作频率明显减少,复查CRP为18.0 mg/L、外周血WBC 5.7×109/L,均较入院时有所下降,一般情况好转出院并于门诊规律随访。随访过程中,患儿秋水仙碱逐渐加量至0.5 mg/d,IL-1、IL-6、TNF-α和粪钙卫蛋白的水平也明显降低,十二指肠溃疡消失(图2),小肠CT中未见明显肠壁增厚(图3)。
2 讨论
以“haploinsufficiency of A 20”、“familial Mediterranean fever”、“autoinflammatory diseases”、“A 20 单倍剂量不足”、“家族性地中海热”以及“自身炎症性疾病”为关键词,年龄限定为0~18岁,国籍限定为中国,分别检索PubMed、万方和CNKI 数据库,共检索到11篇相关文献。其中报道HA20确诊患者11例,中位发病年龄为3.3岁(1个月~7岁9个月),中位确诊年龄8.0岁(1岁3个月~15岁);主要临床表现为周期性口腔溃疡(8/11)、间歇性腹痛(3/11)、不明原因发热和多发性关节炎(10/11)、皮肤损害(4/11)、肺部损害(2/11)、肝脏肿大(2/11)、眼部损害(1/11)。报道FMF 确诊患者34 例,中位发病年龄为5.7 岁(3个月~12岁3个月),中位确诊年龄为9.2岁(1岁9个月~15 岁1 个月);主要临床表现为发热(33/34)、关节痛或关节炎(20/34)、皮肤皮疹(14/34),淋巴结肿大或淋巴结炎(10/34)、腹痛(6/34)、口腔溃疡(4/34)、咽炎(2/34)、贫血(2/34)、胸腔积液(1/34)、肝脾肿大(1/34)[5-15]。没有仅以腹痛为主要临床表现的病例。
在既往文献报道中,反复发生腹痛、腹泻、恶心、呕吐和腹胀是自身炎症性疾病消化道受累的常见表现,又以急性腹痛反复发作最为常见[16]。其中超过95%的FMF患者具有消化道症状,A20单倍剂量不足、TNF 受体相关周期性综合征、高IgD 综合征、要氏综合征也有较常见的消化道症状[17-18]。
HA 20 是一种由TNF-α 诱导蛋白3(tumor necrosis factor alpha-induced protein 3,TNFAIP 3)失功能变异导致的单基因遗传性自身炎症性疾病(OMIM:191163),于2016年首次报道[19]。A20是一种由TNFAIP3基因编码的胞质蛋白,其广泛表达于各型免疫细胞,可抑制核因子κB(nuclear factor-κB,NF-κB)活性和TNF-α 介导的细胞程序性死亡[20]。TNFAIP3基因变异致使其编码蛋白A20对NF-κB通路的负调节作用减弱,从而导致NF-κB 通路介导的促炎细胞因子表达增多,最终表现为HA20。HA20临床表现多变,主要临床表现包括口腔溃疡(60%)、反复发热(44%)、胃肠道损伤(44%)、皮疹(43%)、肌肉骨骼症状(33%)、自身抗体阳性或自身免疫现象(20%)和眼部损伤等[21]。由于HA20早期临床表现可能类似于白塞病、幼年特发性关节炎、系统性红斑狼疮、炎症性肠病等,因此需要临床医师具有足够的甄别意识,对于疑诊患者应尽快完善基因测序检查。
目前HA20的治疗尚无标准方案,主要选择糖皮质激素、免疫抑制剂、生物制剂等,其中糖皮质激素是治疗HA20的首选药物,大多数患者对足量或大剂量糖皮质激素有效。免疫抑制剂包括秋水仙碱、环孢素、甲氨蝶呤、硫唑嘌呤、沙利度胺、霉酚酸酯等。可选用的新型生物制剂包括TNF-α 抑制剂、IL-1 受体拮抗剂、IL-6受体拮抗剂、JAK抑制剂等。此外由于HA20患者体内可以检测到增高的IL-1、TNF、IL-6、IL-18等促炎细胞因子,因此抗细胞因子治疗对抑制全身炎症反应有显著的临床疗效[22-23]。对于严重的药物难治性患儿,造血干细胞移植也可以尝试,但有复发的可能。目前尚未出现有效的靶向治疗药物,因此部分HA20患者的治疗仍存在较大的难度。
FMF是一种遗传性自身炎症性疾病,遗传方式既可以为常染色体显性遗传(OMIM:134610),也可为常染色体隐性遗传(OMIM:249100)[24]。两种遗传方式的致病基因均为MEFV(OMIM:608107),位于16号染色体的短臂,共有10 个外显子。MEFV基因变异可导致其编码的pyrin蛋白数量减少或功能改变,过度活化NALP3炎症小体,从而产生炎症反应。最常见的5个变异位点为M694V、V726A、M680I、M694I和E148Q,其中E148Q在中国人群中变异频率最高。文献报道FMF 常见临床表现依次为:发热、腹膜炎、关节炎、浆膜炎、淀粉样变和非淀粉样肾小球病[25],具有种族和个体差异,且与不同基因变异型相关。目前FMF的诊断多采用Tel Hashomer标准[26],但其对儿童患者特异性不高[27]。有研究者提出并验证了适用于儿童患者的诊断标准,即符合以下5项标准中的2项:①发热(腋下温度>38℃持续6~72小时,发作3次以上);②腹痛(持续6~72 小时,发作3 次以上);③咽痛(持续6~72 小时,发作3 次以上);④滑膜炎(持续6~72小时,发作3次以上);⑤FMF家族史。在临床实践中儿童患者多因反复发热或反复腹痛就诊,特别是急性腹痛患儿由于腹痛剧烈,有时可被误诊为急腹症。
FMF的治疗原则是积极控制发作和炎症反应,预防并发症并改善患儿生活质量及预后。秋水仙碱是治疗FMF的首选,可使60%的患者症状得到控制,或减少其发作频次,重要的是可预防肾脏淀粉样变[28]。诊断明确即应尽早开始秋水仙碱治疗,治疗过程中应每6 个月监测一次。秋水仙碱的不良反应主要包括偶发的肌病和中毒性表皮松解样反应等。如患者发热或者腹痛的症状稳定至少5 年以上,可考虑降低秋水仙碱剂量,但要注意对淀粉样变的随访,对尿蛋白、肾功能以及肝酶进行定期复查[29]。FMF其他治疗药物包括非甾体类抗炎药、糖皮质激素、IL-1 拮抗剂等。对伴有慢性滑膜炎患者,必要时可使用非甾体类抗炎药或腔内注射激素以缓解症状。
猜你喜欢
家庭医药(2023年10期)2023-10-19 19:42:02
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中老年保健(2021年8期)2021-08-24 06:22:06
中国生殖健康(2020年4期)2021-01-18 02:58:10
医学新知(2019年4期)2020-01-02 11:03:52
医学新知(2019年4期)2020-01-02 11:03:52
中国生殖健康(2018年4期)2018-11-06 07:12:16
中国卫生标准管理(2015年6期)2016-01-14 05:17:18
女士(2015年6期)2015-05-30 20:22:32
恋爱婚姻家庭·养生版(2015年2期)2015-05-14 20:45:42
