
一种快速添加或替换蛋白标签的新方法及应用
2021-11-19 08:25张皓刘雪莹钱铭高鸿儒汤超张华王鹏张绍铃吴巨友
南京农业大学学报 2021年6期
张皓,刘雪莹,钱铭,高鸿儒,汤超,张华,王鹏,张绍铃,吴巨友*
(1.南京农业大学园艺学院/江苏省梨工程研究中心,江苏 南京 210095;2.上海农林职业技术学院,上海 201699)
蛋白标签(protein tag)是指利用DNA体外重组技术,与目的蛋白一起融合表达的蛋白或多肽,主要用于目的蛋白的表达、纯化、检测和示踪等[1]。随着功能基因组学和蛋白质组学的快速发展,越来越多的蛋白标签系统被发现,这也极大地丰富了蛋白标签的功能。根据试验的不同目的可以选择不同功能的蛋白标签。目前常用的蛋白标签有谷胱甘肽巯基转移酶(GST)[2]、麦芽糖结合蛋白(MBP)[3]、绿色荧光蛋白(GFP)[4]、用于蛋白纯化和检测设计的八肽(Flag)[5]、六聚组氨酸(6×His)[6]、来源于人c-myc基因的表位标签(Myc)[7]和流感病毒血凝素表位(HA)[8]等。根据蛋白标签相对分子质量的大小被分成2大类:大的蛋白质分子(或蛋白质结构域及其衍生物)和小的多肽片段。前者主要有GST、MBP和GFP等,它们在使用过程中会增加目的蛋白的溶解性,但在蛋白结晶和抗体产生等过程中必须去除标签;后者主要有Flag、6×His、Myc和HA等,多数情况下多肽标签相对分子质量相对较小,对融合蛋白结构的影响较小,不需要从融合蛋白中切除,所以多肽标签更为常用[9]。例如多肽标签Flag和Myc分别由8个氨基酸(DYKDDDDK)和10个氨基酸(EQKLISEEDL)组成的小分子短肽,所以它们对目的蛋白的折叠不会造成明显的影响[10]。6×His标签是由6个组氨酸残基链接上所组成的氨基酸序列,该标签是纯化重组蛋白的首选标签,因为组氨酸残基的序列可以在特定的缓冲液条件下结合到多种类型固定的离子上(比如镍、钴和铜),从而获得易检测和纯化His标签的重组蛋白[11-13]。因此,小分子多肽标签对目的蛋白的结构、功能和生理活性的影响很小,所以在表达及纯化效果方面具有显著的优势。
不同的融合标签系统有其相同点,但各自也有其不同的优势和缺点。融合标签系统受到很多因素制约,例如融合标签系统的纯化条件、融合目的蛋白自身的性质(如等电点和细胞定位等)、纯化的基质及试验材料成本和融合标签的可去除性等[14]。没有任何单一标签可以满足所有试验研究的需要。因此,开发2种甚至多种不同功能蛋白标签的组合使用,现已成为融合标签技术的发展趋势。本研究基于同源重组连接酶反应,利用大肠杆菌转化体系得到同时含有目的基因和所需蛋白标签的重组质粒,为进一步研究目的蛋白的功能奠定了基础。
1 材料与方法
1.1 试验材料
‘砀山酥梨’的花柱,取自南京农业大学梨工程技术研究中心的江浦实验基地。载体pCold-TF、p1300-35S-GFP和pROK2-35S-mCherry由南京农业大学国家梨产业技术研发中心保存。大肠杆菌T1(DH5α)和BL21(DE3)购自北京全式金生物技术有限公司;农杆菌GV3101购自南京百思禾生物科技有限公司。限制性内切酶NdeⅠ、XhoⅠ、XbaⅠ和BglⅡ购自纽英伦生物技术(北京)有限公司;Phanta Max高保真DNA聚合酶、2×Rapid Taq master Mix和同源重组连接酶Exnase Ⅱ 购自南京诺唯赞生物科技有限公司;胶回收试剂盒、质粒小提试剂盒购自爱思进生物技术(杭州)有限公司。
1.2 RNA提取和 cDNA 合成
RNA提取使用成都福际生物技术有限公司多糖多酚植物RNA 提取试剂盒。反转录试剂盒TransScript®One-Step RT-PCR SuperMix购自北京全式金生物技术有限公司。
1.3 引物设计
从梨基因组数据库中下载目的基因S7-RNase的CDS参考序列[15],利用软件Primer Premier 5.0设计目的基因和蛋白标签的引物(表1)。

表1 本研究所用引物及用途Table 1 Primers used in the study and its application
根据同源重组连接酶的原理,将线性化载体和插入片段(插入片段的5′和3′最末端引入线性化载体两端一致的序列15~20 bp)按一定比例混合后,在同源重组连接酶的催化下,37 ℃反应30 min即可完成连接。因此,多标签重组连接酶法需要在目的基因和蛋白标签的正/反向引物5′端前添加15 bp同源臂和酶切位点,用于同源重组连接酶的连接,具体见图1。此外也可以在蛋白标签前添加一些不常用的酶切位点,为后续试验更换目的基因带来便利。设计快速添加目的基因的蛋白标签时,分别为目的基因S7-RNase(去除信号肽)和蛋白标签GFP设计2对引物F1/R1和F2/R2,然后在每个引物的5′端加入相应的15 bp的同源臂和6 bp的酶切位点。例如引物R1与F2之间至少要有15 bp同源臂,引物F1和R2分别与载体pCold-TF被NdeⅠ和XhoⅠ双酶切之后两端要有15 bp同源臂,为了同源重组连接酶的识别、切割特异的重组位点,要连接2个参与重组的分子(表1、图2-A)。同样,设计快速替换目的基因的蛋白标签时,分别为目的基因S7-RNase和蛋白标签mCherry设计2对引物F3/R3和F4/R4,然后在每个引物的5′端加入相应的15 bp的同源臂和6 bp的酶切位点(表1、图2-B)。

图1 目的基因和蛋白标签的引物设计图Fig.1 Primer design diagram of target gene and protein tag

图2 快速添加(A)或替换蛋白标签(B)的流程图Fig.2 Flow chart of the new method of rapid protein tag addition(A)or replacement(B)
1.4 基因克隆及表达载体构建
以‘砀山酥梨’的花柱cDNA和p1300-35S-GFP载体质粒为模板,分别用引物F1/R1和F2/R2经PCR扩增得到目的基因S7-RNase片段1和蛋白标签GFP片段2。pCold-TF空载体用NdeⅠ和XhoⅠ双酶切后,用琼脂糖凝胶回收试剂盒对片段1、2和载体pCold-TF(NdeⅠ和XhoⅠ双酶切)进行胶回收,再将片段1、2和载体pCold-TF(NdeⅠ和XhoⅠ双酶切)在重组酶反应体系下37 ℃反应30 min,最后将样品置于冰上终止反应。重组酶反应体系20 μL:片段1xμL,片段2yμL,双酶切载体片段zμL,5×重组酶反应buffer 4 μL,重组酶 2 μL,用无菌ddH2O补至20 μL。其中,x=[0.04×片段1碱基对数]/片段1的浓度;y=[0.04×片段2碱基对数]/片段2的浓度;z=[0.02×载体片段碱基对数]/双酶切载体的浓度。重组酶反应终止后,立即进行载体转化。从反应体系中取20 μL样品在冰上与100 μL大肠杆菌T1感受态混合,冰浴30 min;在42 ℃水浴锅中热激45 s,立即置于冰上,3 min后在体系中加入700 μL无抗LB培养基,在37 ℃摇床中孵育1 h;将样品涂布于抗性氨苄青霉素(Amp)固体平板,在37 ℃培养箱中倒置培养12 h。从上述的转化平板中挑取10个单克隆菌落分别放入装有200 μL Amp液体LB的2 mL的离心管,在37 ℃摇床中振摇8~10 h。取1 μL菌液为PCR模板,以F5/R5为引物对,进行菌液PCR。PCR体系20 μL:模板 1 μL,正、反向引物各1 μL,2×Rapid Taq master Mix 10 μL,用无菌ddH2O补至20 μL。PCR 产物用琼脂糖凝胶电泳检测。如有目的条带,取100 μL菌液送公司测序。测序结果正确即可得到重组菌株,可以提取质粒进行下一步试验。
以‘砀山酥梨’的花柱cDNA和pROK2-35S-mCherry载体质粒为模板,分别用引物F3/R3和F4/R4经PCR扩增得到目的基因S7-RNase片段3和蛋白标签mCherry片段4。p1300-35S-GFP空载体用XbaⅠ和BglⅡ进行双酶切,其余方法同上。其中菌液PCR的引物为F6/R6,菌落置于含卡那霉素(Kan)的液体LB中摇菌。试验所需引物见表1。
1.5 目的基因S7-RNase的原核表达及亚细胞定位
将去除信号肽的目的基因S7-RNase和蛋白标签GFP构建到His标签的载体pCold-TF中,并转化BL21菌株。采用张皓等[16]的方法,将得到的上述重组表达菌株按1∶50接种到10 mL LB(含100 μg·mL-1Amp)液体培养基中,37 ℃、200 r·min-1振荡培养过夜,活化重组表达菌株。将活化的重组表达菌株按 1∶50 转至15 ml LB培养基(含Amp 100 μg·mL-1)中,37 ℃、200 r·min-1振荡培养至D600为0.5时再置于15 ℃摇床中静置40 min,最后加入终浓度为0.5 mmol·L-1的IPTG,诱导表达24 h。表达完成后取10 mL菌液12 000 r·min-1离心5 min,收集菌体沉淀。沉淀中加入200 μL 0.1 mg·mL-1十二烷基磺酸钠(SDS),混匀后100 ℃水浴10 min,再置于冰上冷却2 min,于4 ℃、12 000 r·min-1离心10 min;取上清液40 μL,加入10 μL 5×蛋白上样缓冲液混匀后,取10 μL进行12%常规SDS-PAGE,考马斯亮蓝染色、脱色后检测重组蛋白表达情况。
将目的基因S7-RNase和蛋白标签mCherry构建到去除GFP标签的载体p1300-35S-GFP上,并将p1300-35S-S7-RNase-mCherry的质粒转入农杆菌(GV3101),经PCR鉴定为阳性后置于28 ℃、200 r·min-1摇床中培养1~2 d。将农杆菌液按体积比为1∶50比例扩繁后5 000 r·min-1离心10 min,收集菌液,将菌液悬浮在含有10 mmol·L-1MES、10 mmol·L-1MgCl2和100 μmol·L-1乙酰丁香酮的诱导剂中,置于室温 50 r·min-1的摇床中孵育4 h。用1 mL去针头无菌注射器吸入菌液,缓缓注射到小叶本氏烟的背面叶片中,侵染完成后置于20~25 ℃的环境下继续生长2 d,使用ZEISS LSM800激光共聚焦显微镜拍照。试验重复3次。亚细胞定位引物见表1。
2 结果与分析
2.1 3种添加或替换目的基因蛋白标签方法用时和步骤的比较
由表2可见:T4DNA连接酶法添加或替换目的基因的1个蛋白标签总用时为4~6 d,而多标签重组连接酶法和融合PCR法总用时2~3 d。通过进一步比较多标签重组连接酶法和融合PCR法,发现融合PCR法在基因克隆步骤上除了扩增目的基因和蛋白标签外,还需要进行预融合PCR和正式融合PCR扩增,因此在基因克隆上融合PCR法用时是多标签重组连接酶法的3倍,可见多标签重组连接酶法在用时方面比融合PCR法还是具备一定的优势,同时这种优势随着扩增片段(目的基因和蛋白标签)长度的增加会进一步扩大。另外,多标签重组连接酶法添加或替换目的基因的1个蛋白标签只需1步即可,而T4DNA连接酶法则需要2步;融合PCR法虽然也只需1步,但是在基因克隆步骤上需要多个步骤。因此,多标签重组连接酶法在用时和步骤上比融合PCR法和T4DNA连接酶法具有更简单、快速和高效等特点。

表2 3种添加或替换目的基因蛋白标签方法用时和步骤的比较Table 2 Time and steps comparison of protein tag addition or replacement of target gene in three methods
2.2 目的基因和蛋白标签片段的克隆及融合载体构建
利用F1/R1和F2/R2引物经PCR扩增得到目的基因S7-RNase(去除信号肽)片段1和蛋白标签GFP片段2。凝胶电泳结果显示,片段1位于500~750 bp,片段2位于500~750 bp,与预期的600和714 bp基本相符(图3-A)。将片段1、2和双酶切载体pCold-TF片段在重组酶Exnase Ⅱ的作用下进行连接,转化和测序。菌液PCR扩增结果可见1 300 bp左右的条带(图3-B),测序结果显示目的基因S7-RNase和蛋白标签GFP已经连接到载体上。同样,利用F3/R3和F4/R4两对引物经PCR扩增得到目的基因S7-RNase片段1和蛋白标签mCherry片段2。凝胶电泳结果显示,片段3位于500~750 bp,片段4位于500~750 bp,与预期的678和711 bp基本相符(图3-C)。将片段3、4和双酶切载体p1300-35S-GFP片段在重组酶Exnase Ⅱ的作用下进行连接,转化和测序。菌液PCR扩增结果可见1 400 bp左右的条带(图3-D),测序结果显示目的基因S7-RNase和蛋白标签mCherry已经连接到载体上。
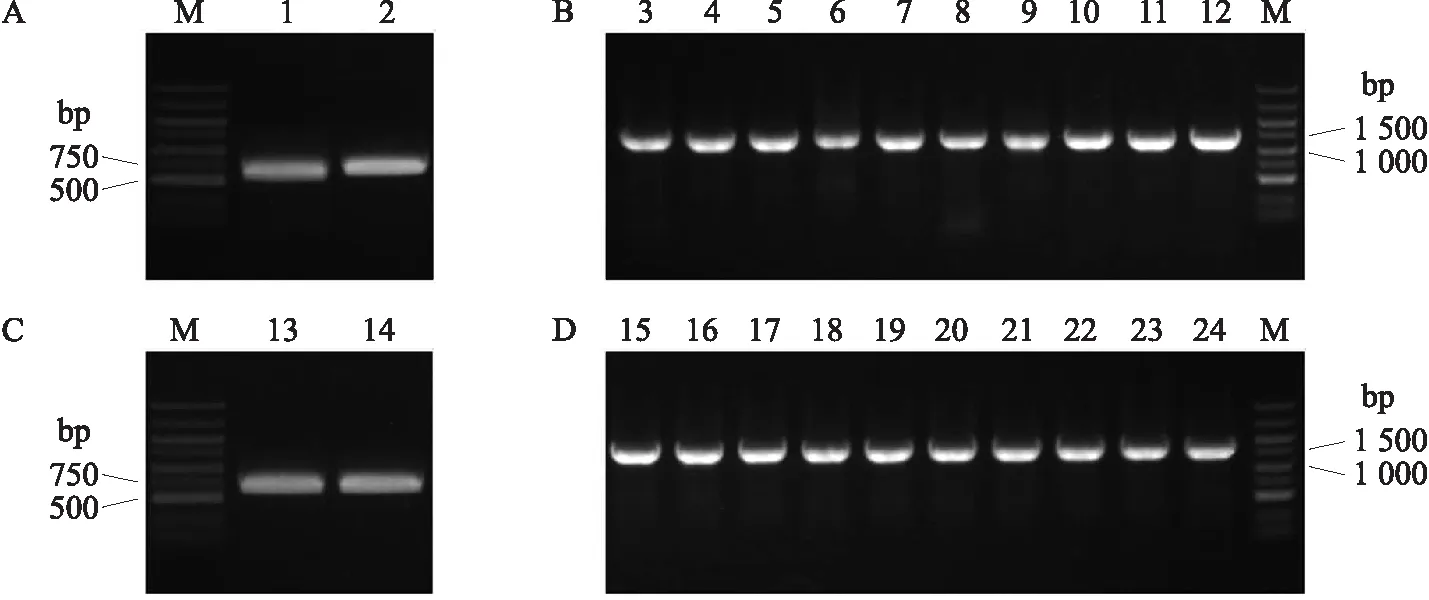
图3 基因克隆(A、C)及阳性克隆的PCR检测(B、D)Fig.3 Gene cloning(A,C)and PCR detection of positive clones(B,D)M. DNA Marker;1~2分别为S7-RNase和GFP蛋白标签的基因克隆;3~12为重组质粒S7-RNase-GFP-pCold-TF转化大肠杆菌的PCR检测;13~14分别为S7-RNase和mCherry蛋白标签的基因克隆;15~24为重组质粒p1300-35S-S7-RNase-mCherry转化大肠杆菌的PCR检测。M. DNA Marker;1-2 indicate gene clones of S7-RNase and GFP protein tag,respectively. 3-12 indicate PCR detection of recombinant plasmid S7-RNase-GFP-pCold-TF was transformed into Escherichia coli. 13-14 indicate gene clones of S7-RNase and mCherry protein tag,respectively. 15-24 indicate PCR detection of recombinant plasmid p1300-35S-S7-RNase-mCherry was transformed into E. coli.
2.3 载体S7-RNase-GFP-pCold-TF重组蛋白的体外表达
将上述重组质粒S7-RNase-GFP-pCold-TF(带有2个蛋白标签:GFP和His)转入大肠杆菌BL21进行原核表达,发现重组质粒S7-RNase-GFP-pCold-TF菌液明显呈浅绿色(图4-A)。通过不同IPTG浓度筛选,最后发现在15 ℃和IPTG终浓度为0.5 mmol·L-1时,重组蛋白表达量最高。S7-RNase(除去信号肽)蛋白相对分子质量约为22×103,带His标签的重组表达载体和GFP标签相对分子质量分别约为50×103和 27×103,所以最终表达的重组蛋白S7-RNase-GFP-His相对分子质量约为100×103,这与SDS-PAGE检测的结果较为一致(图4-B)。
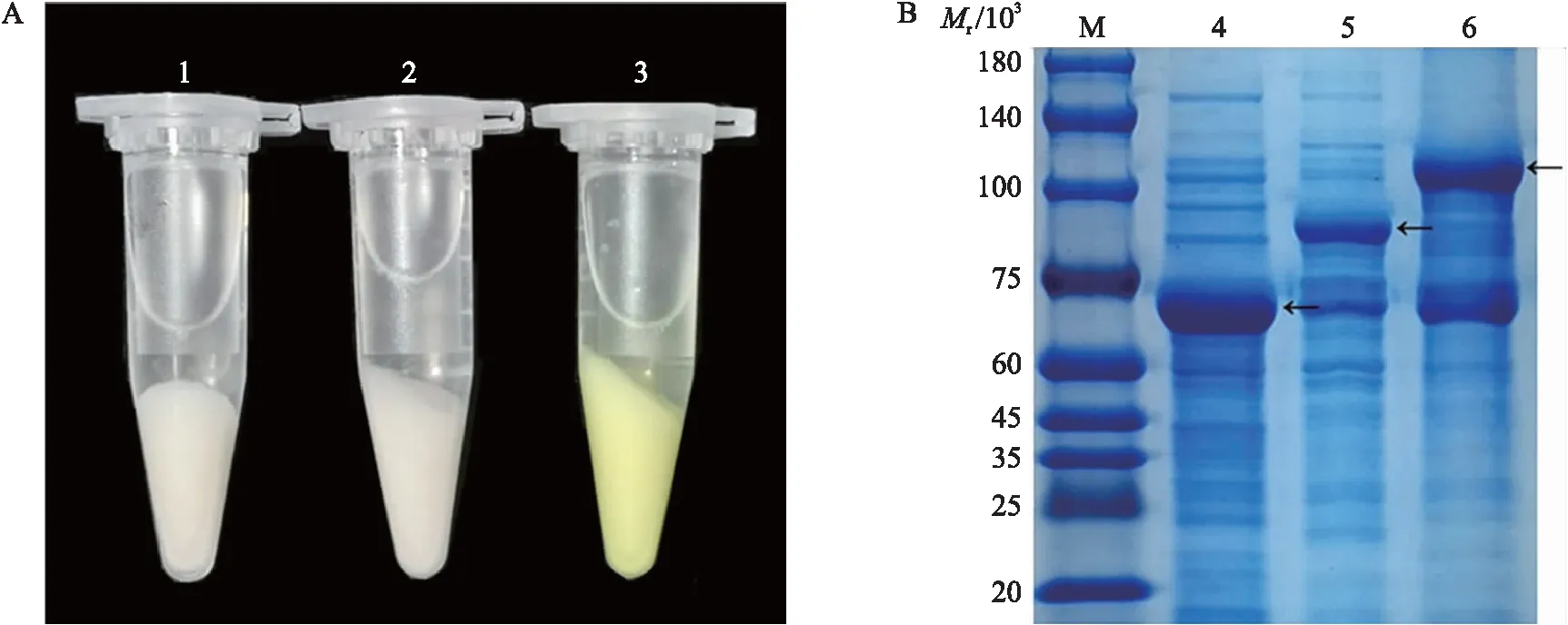
图4 S7-RNase-GFP-pCold-TF在大肠杆菌原核表达系统中的表达(A)及重组蛋白诱导表达后的SDS-PAGE图(B)Fig.4 Expression of S7-RNase-GFP-pCold-TF in prokaryotic system from Escherichia coli(A) and the SDS-PAGE image of recombinant protein after induced expression(B)1~3分别为IPTG诱导后的His、S7-RNase-His和S7-RNase-GFP-His的菌液表达;4~6分别为IPTG诱导后的His、S7-RNase-His和 S7-RNase-GFP-His菌体总蛋白;M. 蛋白质标准分子量。1-3 are the expression from Escherichia coli of His,S7-RNase-His and S7-RNase-GFP-His that are induced by IPTG,respectively. 4-6 are the total protein of His,S7-RNase-His and S7-RNase-GFP-His that are induced by IPTG,respectively. M. The standard of molecular weight of protein.
2.4 目的基因S7-RNase的亚细胞定位分析
如图5所示:GFP空载可在整个烟草叶片的细胞中表达,而S7-RNase-GFP或S7-RNase-mCherry的绿色或红色荧光主要在细胞膜与细胞壁之间有明显积累的趋势;同时在细胞核周围的内质网上也发现了绿色和红色荧光。表明S7-RNase蛋白可在内质网产生并由细胞质运输到胞外。

图5 S7-RNase-GFP和S7-RNase-mCherry蛋白的亚细胞定位Fig.5 Subcellular localizations of S7-RNase-GFP and S7-RNase-mCherry proteinDAPI:4′,6-二脒基-2-苯基吲哚4′,6-diamidino-2-phenylindole. Bar=20 μm.
3 讨论与结论
随着蛋白组学和功能基因组学的兴起,如何获取大量高纯度的蛋白成为一个亟须解决的难题,这也极大促进了融合标签技术和蛋白纯化技术的快速发展。最近,Lu等[17]提出“水稻全基因组蛋白标签计划(RPTP)”的倡议,该计划旨在全基因组范围内让水稻的每一个蛋白编码基因原位标记一个蛋白标签,将极大推动水稻和其他物种的蛋白组学和功能基因组学的发展。此外,通过基因工程技术将具有一定生物学意义的功能基因连接到表达载体中,进行重组蛋白的表达和纯化,有利于进一步研究其具体功能、在细胞内的定位以及与其他蛋白互作等[18]。目前,利用融合标签技术进行目的蛋白的表达、纯化、检测和示踪等方法已得到普遍使用,但其仍然存在一些缺点,如蛋白标签可能干扰与其融合目的蛋白的折叠,从而影响目的蛋白的可溶性和活性,导致其在细胞中不能正常表达和发挥作用[18-19]。因此,合理使用某种蛋白标签对目的蛋白的表达和纯化及其重组蛋白的活性同样也至关重要。
原核表达技术是获得目的蛋白最经济实用的方法之一[20-21]。大肠杆菌因其遗传背景清楚、扩繁周期短、技术操作和培养条件简单等特点,常作为外源蛋白表达的宿主[21]。而GFP作为融合蛋白的表达标签,是一种应用方便、检测手段简单,对融合的目的蛋白产生较少干扰的报告蛋白质[18]。目前在大肠杆菌中利用GFP标签表达外源蛋白是一种常见的技术手段,但T4DNA连接酶法在构建添加或替换目的基因的蛋白标签时存在试验环节多、操作繁杂且转化效率和目的蛋白表达量偏低等问题。尽管融合PCR法为不同来源的任意DNA片段体外连接提供了快速简捷的途径[22],但在基因克隆等步骤上环节较多,因此应用过程也存在一些问题。本研究基于同源重组连接酶反应,利用大肠杆菌转化体系将一个蛋白标签与目的基因同时进行连接从而一步获得试验所需的重组表达载体,同时还可以一步添加多个蛋白标签或替换原有载体上的蛋白标签。该方法具有简单、快速和高效等特点,为进一步研究蛋白结构、功能以及蛋白之间的互相作用等提供了一定的技术支持。
猜你喜欢
今日农业(2022年2期)2022-11-16
种子(2022年6期)2022-08-03
中国糖料(2022年3期)2022-07-04
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
汉字汉语研究(2021年2期)2021-08-30
疯狂英语·新悦读(2020年7期)2020-07-30
中学生物学(2019年7期)2019-10-17
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
湖北农业科学(2017年7期)2017-05-13
