
PLOD2基因变异致Bruck综合征2型临床及基因分析
2021-11-18 08:51郝会民沈凌花张英娴刘晓景卫海燕曹冰燕
临床儿科杂志 2021年11期
郝会民 沈凌花 张英娴 刘晓景 卫海燕 曹冰燕
1.郑州大学附属儿童医院 河南省儿童医院 郑州儿童医院内分泌遗传代谢科(河南郑州 450000);2.国家儿童医学中心 首都医科大学附属北京儿童医院内分泌遗传代谢科(北京 100045)
Bruck综合征(Bruck syndrome)是一种常染色体隐性遗传病,以反复骨折、先天性关节挛缩、身材矮小和翼状胬肉为主要临床特征,由FKBP10或PLOD2基因变异导致,分别命名为Bruck综合征1型(OMIM 259450)和Bruck综合征2型(OMIM 609220),两型的临床表型类似,基因型和表型尚未发现有明显相关 性[1-2]。由于Bruck综合征2 型临床罕见,迄今为止国外文献共报道26例,国内文献尚未见报道,早期诊断和治疗可以改善预后。现回顾分析河南省儿童医院内分泌遗传代谢科确诊的1例Bruck综合征2 型患儿临床及遗传学特点,并通过文献复习,总结Bruck综合征2 型的临床特征及基因变 异特点。
1 临床资料
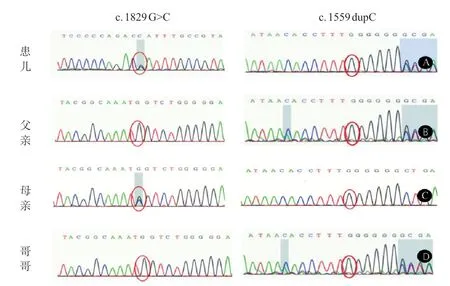
患儿,男,1 岁8 个月,因出生后足外翻及反复骨折,于2020年2月就诊于河南省儿童医院内分泌遗传代谢科。患儿生后即表现为右足外翻,左下肢活动少,并异常哭闹,X 线检查发现为左侧股骨骨折,给予支具固定;6月龄时再次发生左侧股骨骨折,持续支具固定,期间最长去除支具时间约1 周。1 岁4 个月因咳嗽发热行胸部X线检查发现左侧第2、3肋骨骨折、左锁骨骨折,未予特殊处理。此次因左侧股骨骨折再入院。患儿系G 3 P 2,孕36+4周顺产,出生体质量3.05 kg,生后Apgar 评分7 分。母孕期体健。患儿4 月龄抬头,就诊时因佩戴支具尚不会独坐。智力发育无异常。父母非近亲婚配,父母及哥哥表型均正常,否认有家族遗传病史。入院体格检查:身长73 cm( 图1 患儿手足畸形 因患儿反复骨折临床考虑成骨发育不全,行基因检测协助诊断。获得患儿父母知情同意并签署书面同意书,且经医院伦理审查委员会批准(2021-K-023),抽取患儿及父母、哥哥外周血样各2 mL,送武汉康圣达医学检验所检测。全外显子测序显示PLOD 2基因存在编码区第17 外显子c.1829 G>C(p.Trp 610 Ser)和第13a外显子区域c.1559dupC(p.Val523CysfsX7)复合杂合变异,分别来源于母亲和父亲,哥哥携带c.1559 dupC 杂合变异(图2)。参考美国医学遗传学和基因组学协会(American College of Medical Genetics and Genomics,ACMG)基因变异解读指南,c.1559dupC变异为罕见变异,在千人基因组、EXAC、gnomAD 数据库中没有收录,且为HGMD 数据库已报道的致病变异,导致第521 位的氨基酸赖氨酸发生变异,随后的氨基酸发生移码,可能使蛋白结构不能行使正常功能,评级为致病性(PVS 1+PM 2+PP 4);c.1829 G>C 为未报道过的变异位点,参考ACMG基因变异解读指南,该变异位点无人群携带率,检测到复合杂合致病变异位点,SIFT和Ployphen软件提示该变异位点会对基因或基因产物造成有害影响,临床症状符合,评级为可能致病(PM2+PM3+PP3+PP4)。 图2 PLOD2基因测序图 治疗上予口服钙剂(元素钙500 mg/d)、维生素D(400 IU/d);住院期间静脉用唑来膦酸钠(0.1 mg/kg)治疗1次,6个月后重复应用1次,目前共应用2次,以后计划每年1 次;继续支具固定治疗3个月。每3 个月随访1次,至今随访1年。1岁11个月时身高75 cm,体质量9.2 kg,去除支具,会扶走,复查左股骨侧位片骨质较前明显好转;2岁8个月身高79 cm,体质量 11 kg,能独走,未再发生骨折。 PLOD 2基因包含20个外显子,长度为91 745 个碱基,编码758个氨基酸。PLOD2基因可编码 3q23-24染色体上的端肽赖氨酸羟化酶2(LH2),LH2可以羟基化I型胶原蛋白氨基末端肽和羧基末端肽中的赖氨酸残基,这些残基对骨细胞外基质交联的形成至关重要,PLOD 2基因变异导致胶原末端肽的赖氨酸残基羧基化程度缺陷,从而增加骨的脆性,导致骨折发 生[3]。胶原端肽与螺旋结构域之间的分子间共价交联对胶原纤维强度至关重要,骨、软骨、韧带、肌腱等组织交联主要通过羟基化途径形成,因此,PLOD2变异可导致韧带、肌腱和支持组织胶原交联化学改变,引起关节挛缩。 以“PLOD2”、“Bruck综合征”、“Bruck syndrome”为关键词在中国知网和万方数据库从建库至2020 年12 月查阅文献,未检索到符合条件的中文文献,国外文献共报道了26例患者,结合本例共27例患者。其中男16例,女11例,2例胎儿期夭折。相关表型依据出现频次排列:多发骨折27例(100%),先天性关节挛缩17例(62.9%),脊柱侧凸16例(59.2%),手指屈曲12例(44.4%),畸形足12例(44.4%),身材矮小10例(37%),蓝巩膜9例(33.3%),翼状胬肉6例(22.2%)。27例中有类似家族史12例(44.4%);监测骨密度10例(37.0%);接受膦酸盐治疗11例(40.7%),最长应用时间5 年。27例患儿均进行了基因检测,发现PLOD2基因变异,共发现24种不同的变异位点,分别分布在4、5、8、11、12、13a、14、16、17、18、19外显子和4、12、18内含子中,主要集中在17外显子,包括错义变异、无义变异、缺失、重复和剪接位点变异,17例为纯合变异,10例为复合杂合变异,均符合常染色体隐性遗传。其中c.1856G>A(3例)和c.1559dupC(3例)可能为热点变异[1-7]。 Bruck综合征2 型临床特点为先天性关节挛缩、弥漫性骨质疏松、婴儿期或儿童早期骨骼脆弱易骨折、身材矮小、重度肢体畸形、进行性脊柱侧凸和翼状胬肉。骨折多发生于婴儿或儿童早期,部分患儿于出生时因分娩创伤即发生骨折,每年3~6 次,严重者可达10余次,骨折通常发生在上肢和下肢骨干,包括尺骨、股骨、胫骨、腓骨等,上肢和下肢畸形是由反复骨折引起。因此Santana 等[4]提出了生长期间疾病的模式:出生时的挛缩、生长期间的骨折和随后的畸形。在国内指南归类于成骨发育不全中,与成骨不全表现不同的是,Bruck综合征2型有先天性关节挛缩和翼状胬肉,没有听力下降、关节韧带松弛、牙本质发育不全和心脏瓣膜病变等表现,且成骨不全多有骨折阳性家族史[8]。目前Bruck综合征2型尚未见报道与语言及认知发育迟滞有关[9]。 目前尚无针对致病基因变异的有效治疗方法,非手术治疗方法与成骨不全相同,旨在增加患者的骨密度、降低骨折率、改善骨畸形,提高患者生活质量,需要骨科、康复科、内分泌科等多学科共同协作。药物治疗选择也是从治疗其他代谢性骨病方法中借鉴,通过应用膦酸盐抗骨吸收和促进骨合成代谢从而增加骨量,降低骨折风险[5,10-12]。一项应用唑来膦酸盐治疗Bruck综合征2 年的前瞻性研究表明,应用剂量为0.1 mg/kg,每6个月1次,应用6个月后脊柱和髋关节BMD和Z值评分提高,骨折率显著下降,行走能力得到改善,应用2 年后改善更为明显,且患者对该药具有良好的耐受性,27例患者中,接受磷酸盐治疗11例,最长治疗时间5 年,末次监测骨密度基本正常,取得了较好疗效[10]。髓内钉的应用可以减少骨折次数及维持长骨功能;物理治疗和矫正器的使用有助于阻止挛缩的进展,有益于患者日常活动;在药物降低骨折风险同时配合手术矫正畸形,改善负重力线,通过脊柱融合和内固定治疗脊柱畸形[13-14],可提高患儿生活质量。PLOD2变异患者合并有骨密度降低,因此本例患儿给予补充维生素D和钙剂[2,15],住院期间静脉用唑来膦酸钠(0.1 mg/kg)治疗1 次,6 个月后重复应用1次,目前共应用2次,以后计划每年1 次,继续支具固定治疗3个月。患儿每3个月随访1次,至今随访1年,1岁11个月时身高75 cm,体质量9.2 kg,去除支具,会扶走,复查左股骨侧位片骨质较前明显好转;2 岁8个月时身高79 cm,体质量11 kg,能独走,未再发生骨折。 Bruck综合征2型因常合并严重的双侧关节挛缩,且伴有翼状胬肉,关节功能和骨折愈合将会明显受影响,且会增加骨折发生率,预后往往比成骨不全更差,但如果挛缩程度较轻,仅从骨折类型上难于与成骨不全相鉴别,应积极完善基因检测明确诊断。27例中44.4%(12/27)有类似家族史,因此对于有阳性家族史者,产前诊断至关重要。 本研究显示,Bruck综合征2 型患者骨折发生较早,但患者第一次就诊年龄往往迟于骨折发生后2~3年,明确诊断年龄甚至更晚。报道病例中临床表现、实验室数据未能详细描述,但也充分说明了临床医师对疾病缺乏认识,因此对于反复多发骨折,合并先天性关节挛缩、手指屈曲、畸形足、脊柱侧凸、蓝巩膜和翼状胬肉等表现患者,应警惕Bruck综合征2型的发生,尽早完善基因检测明确诊断,尽早干预治疗可改善预后。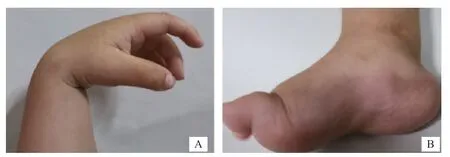
2 讨论
猜你喜欢
中日友好医院学报(2022年1期)2022-11-28
按摩与康复医学(2022年23期)2022-11-25
技术与创新管理(2022年3期)2022-05-31
天津医药(2021年6期)2021-12-08
健康体检与管理(2021年7期)2021-09-19
云南医药(2021年3期)2021-07-21
医学信息(2020年4期)2020-04-09
中老年健康(2016年9期)2016-11-18
中国民族民间医药·下半月(2014年2期)2014-09-26
中国当代医药(2014年17期)2014-09-12
