
GLB1基因变异致GM1神经节苷脂贮积症Ⅱ型1例报告并文献复习
2021-11-18 08:51李甜馨陆相朋彭琰琰卢婷婷廉文君都修波马丙祥
临床儿科杂志 2021年11期
李甜馨 陆相朋 彭琰琰 卢婷婷 廉文君 冯 斌 都修波 马丙祥 郑 宏
1.河南中医药大学;2.河南中医药大学第一附属医院儿科(河南郑州 450000)
GM 1神经节苷脂贮积症(GM 1 gangliosidosis)是因β-半乳糖苷酶基因(GLB 1)变异,导致以 GM 1神经节苷脂代谢异常为主的一种先天性遗传代谢病,属于溶酶体贮积症,为常染色体隐性遗 传病[1]。GM1神经节苷脂贮积症可根据临床表现,从重至轻分为Ⅰ型(婴儿型),Ⅱ型(晚期婴儿型、少年型),Ⅲ型(成人/慢性型),总发病率为1/300 000~ 1/10 000。我国尚无明确发病率报道,婴儿型及晚期婴儿型为我国常见表型[1-2]。现报告1例GM 1神经节苷脂贮积症Ⅱ型患儿,并结合相关文献探讨我国GM 1神经节苷脂贮积症Ⅱ型患儿的临床特点及基因型。
1 临床资料
患儿,男,1岁2个月,于10月龄患肺炎后发现运动发育倒退,不能独坐、不会爬、不能独站。当地医院发现患儿肌张力低下,运动发育评估示相当于7 月龄水平,未予治疗。患儿系G3P3,母孕期无异常,足月顺产,出生体质量3.5 kg,身长52 cm,出生时无缺氧窒息、病理性黄疸史,母乳喂养,10月龄前生长发育基本同正常同龄儿。父母体健,非近亲婚配。
入院体格检查:身长74 cm(P3~P10),体质量10.5 kg(P50~P85),头围45.5 cm(P15~P50),前囟已闭,表观无明显异常,心肺腹无异常,四肢脊柱关节未见畸形;颅神经检查不配合;追视、追听可,独坐不稳、不会爬,四肢肌张力低下,双侧膝腱反射可引出,病理征阴性。实验室检查:血、尿常规无明显异常,丙氨酸氨基转移酶(ALT)13.2 U/L,天冬氨酸氨基转移酶(AST)105.4 U/L,碱性磷酸酶180.6 U/L,肌酸激酶46.4 U/L,肌酸激酶同工酶23.3 U/L,乳酸脱氢酶497.9 U/L,低密度脂蛋白98.5 U/L,α-羟基丁酸脱氢酶332.3 U/L;尿有机酸分析提示营养障碍(草酸、3-羟基丙酸、丙二酸、甘油酸、3-甲基戊烯二酸、3-羟基戊二酸浓度增高);血氨基酸及酯酰肉碱谱分析提示乙酰肉碱38.45 µmol/L,鸟氨酸48.18 µmol/L。肝胆脾胰及心脏彩超、左手正位及脊椎正位X线未见明显异常。脊椎侧位X 线可见鸟嘴突样改变(图1)。脑电图未见异常放电。头颅磁共振成像(MRI):双侧侧脑室三角区旁异常信号,考虑髓鞘化终末带、髓鞘化发育不良。Gesell发育评估:适应能力64.2分,粗大运动58.6分,精细动作71.5分,言语能力58.4分,社交能力57.1分,综合发育商61.9分,整体发育迟滞,适应能力、粗大运动、精细动作、言语能力和社交能力均属轻度缺陷。
为进一步明确诊断,经河南中医药大学第一附属医院医学伦理委员会批准,患儿家属知情同意后,采集患儿及其父母静脉血2 mL,提取外周血白细胞全基因组DNA,构建基因组文库,采用芯片捕获全外显子及相邻内含子区域,并富集目的基因片段,通过Illumina Hiseq系列测序仪进行基因测序(北京福佑龙惠遗传病诊所),目标序列测序覆盖度不低于99%,疑似致病变异验证对目标序列进行PCR 后行Sanger 测序验证。结果显示,患儿在GLB1基因存在2处杂合变异(图2):Exon2,c.202C>T,p.R68W,来自母亲,已报道致病变异;Exon8,c.832G>A,p.G278S,来自父亲,是GLB1基因第8外显子的832号处的G碱基变异为A 碱基,使得GLB 1蛋白278 号密码子由甘氨酸变异为丝氨酸。
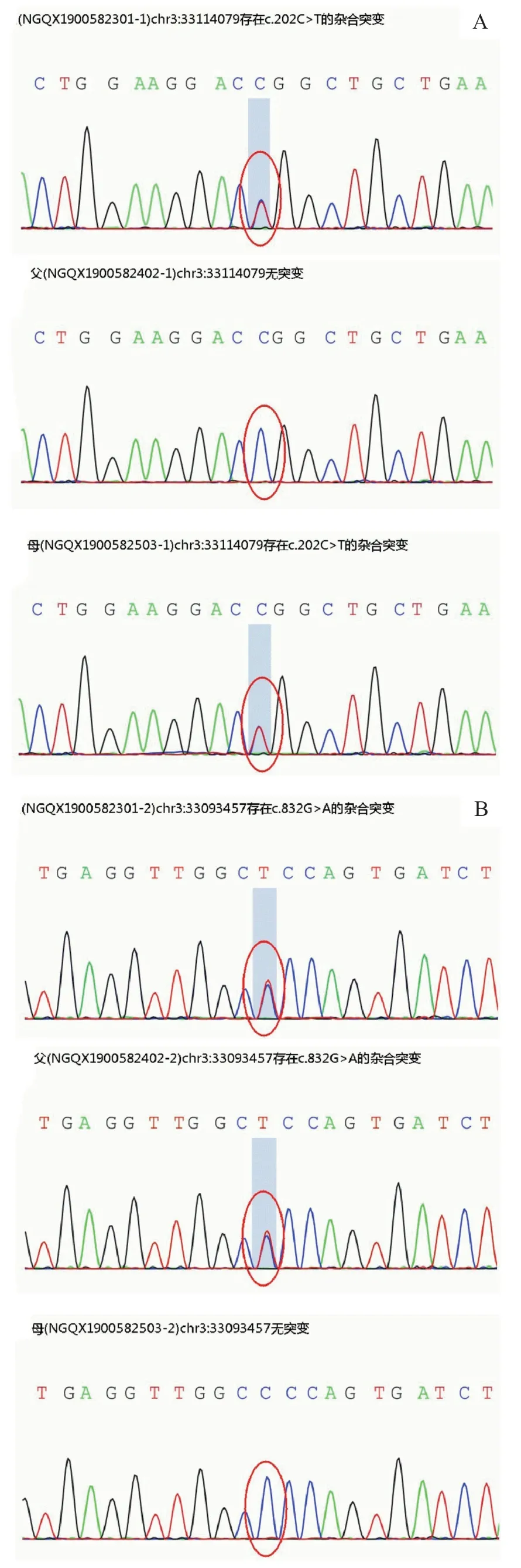
图2 患儿及父母GLB1基因测序图
经查阅千人基因组、千人南方、千人北方等数据库,均未见收录和报道GLB1基因的c.832G>A。根据美国医学遗传学与基因组学学会(American College of Medical Genetics,ACMG)变异解读指南[3]进行生物信息学致病等级分析,c.832 G>A 属于错义变异,为可疑致病变异(PM 1+PM 2+PM 3+PP 3):错义变异位于深入研究的无良性变异的外显子功能域(PM1);所有正常人群数据库频率<0.005,属于低频变异(PM 2);该变异反式位置上检测到致病/疑似致病变异(PM3);多种统计方法预测出变异对基因(基因产物)有影响,保守性及蛋白结构预测有害(PP3)。其中Sift预测结果为 damaging(0.009),Polyphen2_HDIV为probably damaging(1.0),Polyphen 2_HVAR 为probably damaging(0.998),PROVEAN为deleterious(-5.45),MutationTaster为disease_causing(1),M-CAP为damaging(0.489507),REVEL为deleterious(0.939),GERP为D(5.42),剪接预测(GTAG)有害。此外,蛋白质二级结构预测(predictprotein、Phyre2)结果显示,变异对α螺旋结构有一定影响。
综合分析显示,患儿属于复合杂合变异,符合常染色体隐性遗传规律,提示为GM 1神经节苷脂贮积症Ⅱ型。
患儿确诊后,予口服药物对症治疗,以及运动训练、物理治疗等综合康复治疗。随访至2岁2个月时运动、言语、认知进一步倒退,不会独站、独坐,交流存在障碍,全面发育落后于正常同龄儿,并且出现吞咽困难。
2 讨论
以“GM 1神经节苷脂贮积症Ⅱ型”、“GLB 1基因”为检索词,在万方数据库、中国知网数据库、维普数据库检索,以“GLB1AND Chinese”、“GLB1AND China”为检索词在PubMed 数据库检索,经检索、筛选,共得到8 篇文献[1,4-10],其中中文文献5 篇,外文文献3篇。加本例患儿,共有17例中国患儿。GM 1神经节苷脂贮积症Ⅱ型中国患儿的基因变异类型包括错义变异、移码变异,位点c.446C>T变异较多,其次为c.495_497delTCT、c.710A>G、c.1979_1980insG、c.146 G>A。见表1。此类患儿均可见运动、智力的发育倒退,除此之外的主要临床表现为肌张力高(12/17)、癫痫(11/17)、斜视(7/17),实验室检查可见AST升高伴ALT正常(8/17),颅脑MRI多见髓鞘化发育不良(6/17)。少数可见巴氏征阳性、步态异常、大头畸形、瞳孔无光反射/反射迟钝、反复肺部感染、面部表观异常、肝脾肿大、脊柱侧凸、眼底发灰、苍白球T2 低信号、骨发育不良、脊椎侧位X 线可见鸟嘴突样改变、腹胀。
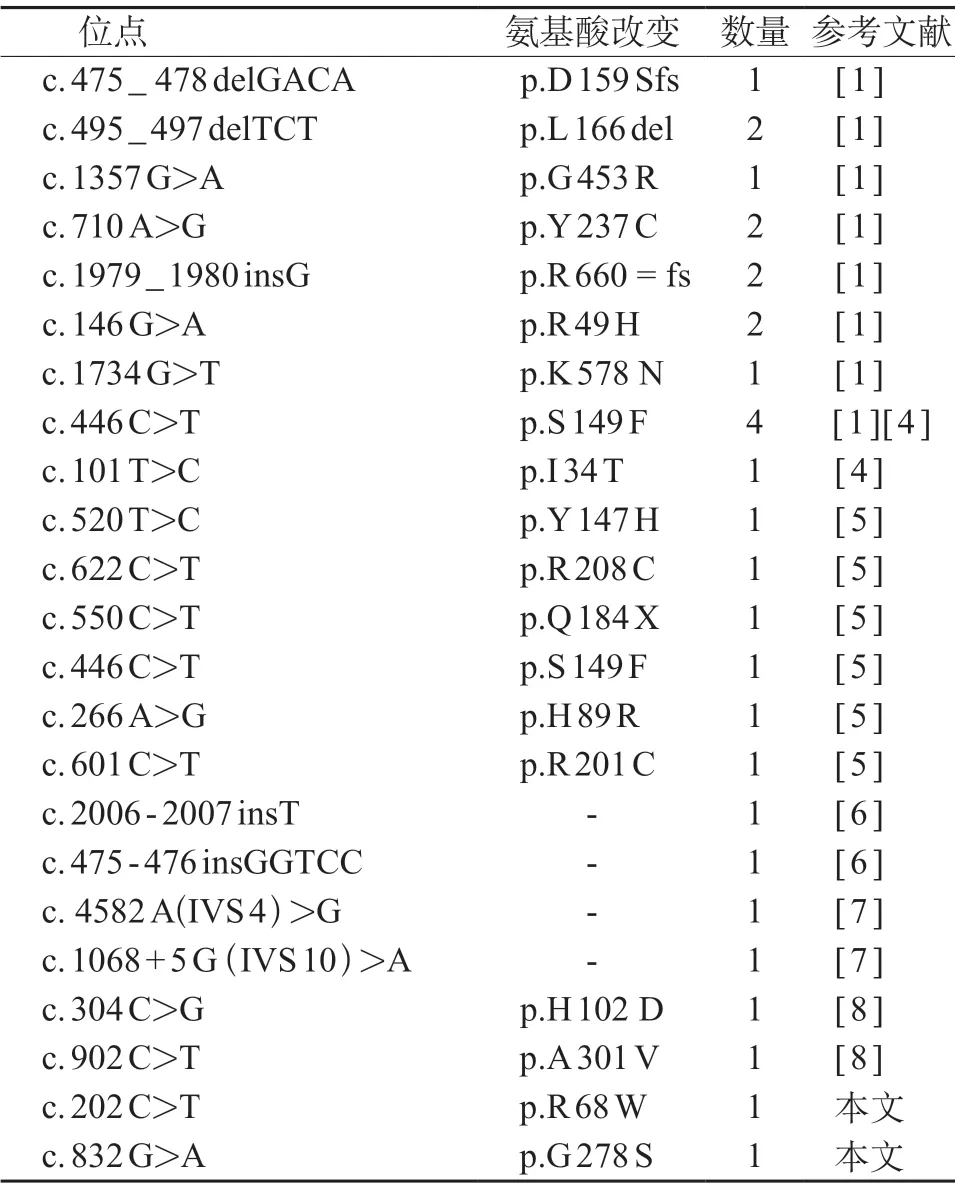
表1 我国GM1神经节苷脂贮积症Ⅱ型患儿 GLB1基因变异情况
GM 1神经节苷脂贮积症是由GLB 1基因变异所致的常染色体隐性遗传病,可影响多脏器病变,是GM1神经节苷脂沉积导致的一种神经退行性疾病[11]。GM1神经节苷脂贮积症在临床并无严格的分型,通常根据严重程度分为3型,有助于了解发病时间、症状和进展速度的变化[12]。
GM 1神经节苷脂贮积症Ⅱ型表现为于6 月龄后起病,运动及认知功能减退、角膜混浊、共济失调、骨骼异常、斜视和癫痫发作风险增加,颅脑MRI 可见进行性脑萎缩、苍白球低信号、壳核高信号和铁沉 积[12-13]。然而,GM1神经节苷脂贮积症各型的表现存在部分重叠,都可能存在认知能力减退、肌张力低下、癫痫、共济失调。颅脑MRI可见弥漫性脑萎缩和脑白质异常,白质T2加权相高信号;丘脑T1高信号和T2低信号;基底核及苍白球T 2 加权相低信号,壳核的T 2 加权相高信号;常见致死原因为吸入性肺炎和心肌病[4,14]。我国患儿临床表型与上述相符,但有报道1例Ⅱ型患儿于2岁2个月时死于严重消瘦和低血容量性休克,这可能与我国患儿多有报道的咀嚼困难/饮水呛咳导致喂养困难有关[8]。本例患儿就诊时体格、营养方面未见明显异常,与既往我国文献报道相符,但随访至2岁2个月时,患儿出现喂养困难,与疾病进展相符,应进一步关注其营养状况,必要时给予鼻饲管喂养。
除此之外,既往报道GM 1神经节苷脂贮积症Ⅱ型患儿最初的特征通常是神经和骨骼的异常,在语言上可达到10~20个单词的词汇量,但直到发病前都难以将单词组合成句;运动、言语等技能在12~18个月时达到平台期,可能会学习站立,但很快就会倒退或丧失,2岁前从未实现独走,独坐能力逐渐退化;骨骼方面存在复合性骨发育不良,虽短管状骨未受影响,但可影响尚未发育成熟的骨骼,并伴有迟发性骨化征象(如齿状突发育不全、卵圆形椎体、髋臼顶部倾斜的髂体发育不良和股骨头骨骺异常)[1,15]。文献复习发现,我国患儿均报道有智力、运动倒退,但大多未见骨骼异常;临床表现为神经系统异常,以肌张力增高、癫痫、斜视为主;少数可表现为肌张力减低、声音诱发的惊跳反应、眼球震颤等。本例患儿无癫痫发作,脑电图无异常,但不会独坐、不会独站、四肢肌张力低下,适应能力、粗大运动、精细动作、言语能力和社交能力均存在缺陷;脊椎侧位X线可见鸟嘴突样改变。随访至2岁2个月,患儿运动、言语、认知能力进一步倒退,未实现独走,不会独坐,交流存在障碍,最终结合基因检测,诊断为GM1神经节苷脂贮积症Ⅱ型。
本例患儿基因检测发现2 个变异位点,其中c.202 C>T 在2003 年报道该位点变异不会干扰GLB 1基因转录和翻译,但可以使β-半乳糖苷酶失活[14],其可能的致病机制是该位点变异导致β-半乳糖苷酶前体加工异常,异常的β-半乳糖苷酶无法运输至溶酶体,β-半乳糖苷酶无法正常形成功能复合物并与保护蛋白/组织蛋白酶A(proteetive protein/ cathepsin A,PPCA)组装,影响神经酰胺酶(neuraminidase 1,NEU1)、N-乙酰半乳糖胺-6-硫酸酯酶(N-acetygalactosamine-6-sulfate,GALNS)和/或变异酶的动力学特性及活性。其中PPCA、NEU 1 与β-半乳糖苷酶共同形成溶酶体复合物,协同分解复杂大分子,其相互作用至关重要,GALNS也可能参与此过程[17]。而β-半乳糖苷酶活性缺乏会使GM 1神经节苷脂及相应的去唾液酸衍生物GA 1 在外周及中枢神经系统的神经元等器官和组织中积累,毒性底物积聚造成的进行性损害导致脑神经元破坏,可表现为中枢神经系统局部小胶质细胞中GM1神经节苷脂积聚,进而导致这些细胞内炎症细胞的活化和浸润增加,引起神经系统病变[18-19]。对1例GM1神经节苷脂贮积症Ⅱ型日本女性患者的尸检证实此其脑部严重萎缩(主要见于额叶),并在显微镜下发现,大脑皮质、壳核、小脑等部位有严重的胶质增生和神经元丢失,后者包括浦肯野细胞层和颗粒细胞层,而海马基本维持原状;这例患者病程较长,在人工呼吸支持下于40岁死于肺炎[20]。本例患儿肺炎后出现运动倒退。有报道1例患儿腹胀后出现发育倒退并存在反复肺部感 染[8],1例患儿出现与上呼吸道感染有关的高热惊厥、癫痫[10],故考虑感染为GM 1神经节苷脂贮积症II 型的高危因素。
本例患儿另一位点c.832 G>A 是GLB 1基因第8外显子的832号处的G碱基变异为A碱基,使得GLB1蛋白278 号密码子由甘氨酸变异为丝氨酸,尚未收录报道。尽管ACMG指南变异解读指南判定该位点为可疑致病变异,但患儿有较为明显的临床表现,统计方法预测出变异对基因(基因产物)有影响。家系sanger测序验证结果显示此双杂合变异分别来自于其父母,为复合杂合变异。综合患儿的基因检测结果及临床表型,可基本确定该位点与c.202C>T杂合变异为患儿的致病性变异。一般来说,变异核苷酸的位置和性质可决定β-半乳糖苷酶的残余活性,从而预测个体症状的轻重以及发病年龄的早晚,但该病是否存在基因型-表型尚存争议[19,21]。
迄今为止,GM1神经节苷脂沉积症的治疗方法尚不成熟、治疗费用昂贵,患者确诊后多为对症治疗,故随访和长期管理至关重要,如定期监测脑电图,一旦发现癫痫发作应及时对症处理,密切观察心脏、骨骼和眼睛,并建议接受常规免疫接种,尤其是流感和肺炎球菌疫苗[2]。出现喂养困难时,考虑尽早开始胃管饲养[14]。GM1神经节苷脂贮积症预后不良,患儿多数无明显表观异常,病理征(克氏、布氏和巴氏征)可能阴性。新生儿筛查检测酶活性可能由于人群中存在假缺失基因变异而导致假阳性[22],故当患儿出现明显运动、智力倒退时,有必要进行基因检测。
本例GM 1神经节苷脂贮积症Ⅱ型患儿,扩大了我国患儿的临床表型及基因变异谱。
猜你喜欢
介入放射学杂志(2022年8期)2022-11-26
中草药(2022年20期)2022-11-15
食品科学(2022年20期)2022-10-31
现代食品科技(2022年9期)2022-10-09
中草药(2022年17期)2022-09-05
云南中医中药杂志(2022年5期)2022-05-18
心电与循环(2021年4期)2021-11-29
健康之家(2021年19期)2021-05-23
锻压装备与制造技术(2020年6期)2021-01-25
宝藏(2020年3期)2020-10-14
