
GTPBP3基因变异相关线粒体病1例报告并文献复习
2021-11-18 08:51符跃强
临床儿科杂志 2021年11期
杨 倩 符跃强
重庆医科大学附属儿童医院重症医学科 儿童发育疾病研究教育部重点实验室 国家儿童健康与疾病临床医学研究中心 儿童发育重大疾病国家国际科技合作基地 儿科学重庆市重点实验室(重庆 400014)
原发性线粒体病是由线粒体基因或核基因缺陷导致的线粒体功能障碍或结构异常而引发,常致多系统受累,是最为常见的先天性代谢缺陷,其临床表现及遗传方式具有高度的异质性,致死及致残率高。三磷酸鸟苷结合蛋白3(guanosine triphosphate -binding protein 3,GTPBP3)基因是线粒体病的致病基因之一。国外关于GTPBP3基因相关线粒体病报道极少,国内尚未见报道。本文回顾分析1例诊断明确的GTPBP3基因变异相关的线粒体病女性患儿的临床特征及基因变异特点,并对相关文献进行复习。
1 临床资料
患儿,女,3岁4月龄,因面色苍白、呼吸困难3小时,惊厥1 次入院。入院前3 小时患儿于外院行康复治疗时突然出现呼吸困难、面色发绀,伴惊厥发作1次,主要表现为双目凝视,呼之不应,即予甘露醇、吸痰和止惊等处理,气管插管后转至重庆医科大学附属儿童医院,以惊厥待查、重症肺炎、肺出血可能收入院。患儿系G2P2,足月剖宫产,出生体质量3 450 g,无窒息抢救史。母孕期无异常,无放射线接触史。患儿1 岁前生长发育和同龄人无明显差异,1 岁后生长发育比同龄人落后;1 岁半扶走,2 岁独立行走,3 岁半能跑跳,但动作较缓慢;2 岁叫爸爸妈妈,3 岁半才能听懂他人简单话语,目前自我表达能力欠佳,不能完整表述简单句。有一哥哥,14 岁,体健,生长发育、语言及智力情况与同龄人无差异。否认家族性遗传病史,父母非近亲婚配。入院体格检查:体温37.5℃,呼吸30 次/min(机械通气),心率150 次/min,血压 105/52 mmHg,氧饱和度98%,体质量14.0 kg;发育迟滞,营养良好,精神欠佳,深度昏迷,面色欠佳,无脱水貌;全身无皮疹,无牛奶咖啡斑;心、肺、腹无异常。实验室检查:血气分析pH 6.99,pCO232.7 mmHg,HCO3
-7.9 mmol/L,TCO28.9 mmol/L,SBC 8.8 mmol/ L,ABE -24 mmol/L,乳酸29 mmol/L,血糖19.1 mmol/ L;超敏肌钙蛋白I 1.91 μg/L,肌酸激酶MB 同工酶15.92 μg/L;丙酮酸599 μmol/L,β-羟丁酸0.71 mmol/L,游离脂肪酸0.69 mmol/L;血氨 45.1 μmol/L。入院前头颅磁共振(MRI)示双侧丘脑对称斑片状T1低、T2高信号影,T2FLAIR及DWI呈高信号(图1A、B);桥脑膨大,呈T1稍低、T2稍高信号影,FLAIR及DWI呈稍高信号影,双侧小脑齿状核可见斑片状T1低、T2高信号影,FLAIR及DWI呈稍高信号影(图1C、D)。入院后超声心动图示左室稍增大,左室后壁动度降低;左肺动脉血液流速稍增快;左心收缩功能降低,舒张功能无明显异常,射血分数44%。心电图示窦性心律。脑电图示界限性,清醒闭眼时双侧后头部呈5~7 Hz θ节律为主,枕区波幅最高,夹杂少量中幅δ波及极低幅快波,双侧基本对称;闪光刺激时未见间隙性光刺激相关异常放电;睡眠波及睡眠周期基本正常,可见双侧基本对称的顶尖波、睡眠纺锤波、K-综合波;监测中未见明显发作性脑电图改变。
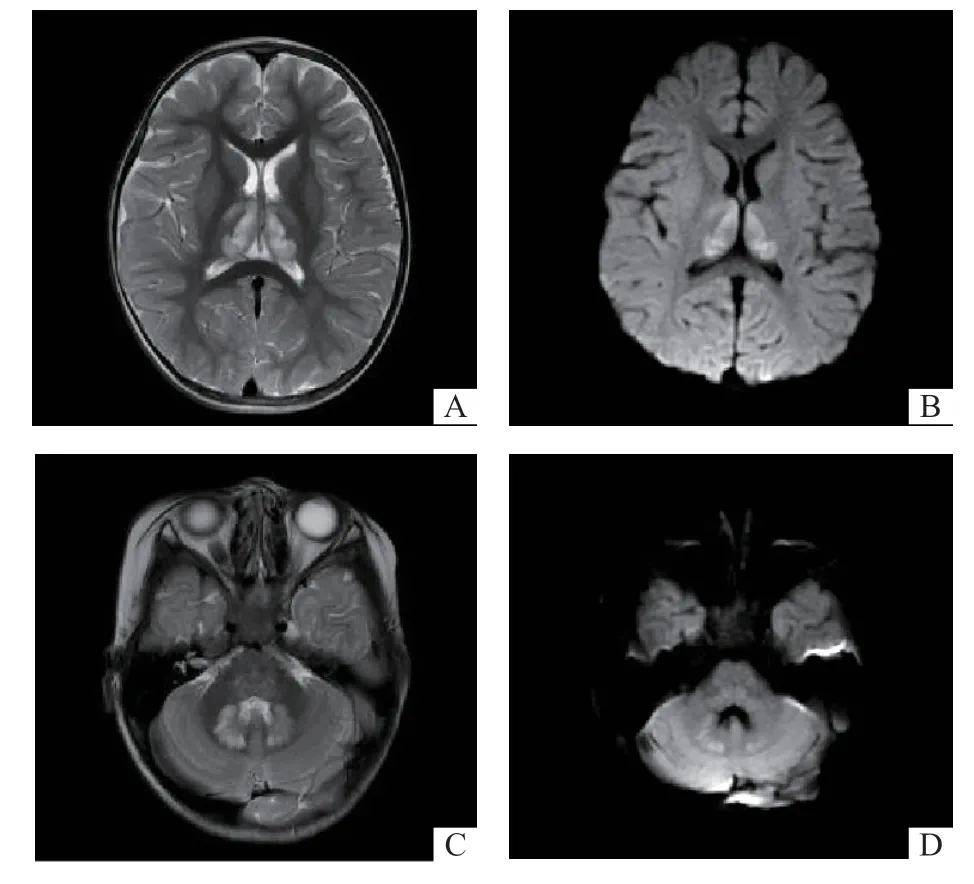
图1 患儿MRI 表现
入院后患儿行呼吸机辅助通气,出现难以纠正的严重代谢性酸中毒、高乳酸血症,并伴有高钠血症。在多普勒超声引导下行颈内静脉穿刺置管术,并行持续有创血压监测。入院后行血液透析治疗,无血液透析滤过相关并发症。血液透析后,血气明显好转。同时予B族维生素、辅酶Q10、左卡尼汀等治疗10天后,病情好转,神志清楚,生命体征平稳,出院。
患儿有全面性生长发育迟缓,伴严重代谢性酸中毒、高乳酸血症及神经系统症状,结合头颅MRI,考虑遗传代谢性疾病可能。经医学伦理审核,并取得患儿父母知情同意后,行基因检测。抽取患儿及其父母外周静脉血2 mL,使用 Solpure Blood DNA Kit试剂盒(Magen 公司)提取基因组DNA,取1 μg基因组DNA样本经Q800R超声破碎仪处理后得到约350 bp的DNA 片段,制备测序文库,用特异性捕获探针,对OMIM收录的遗传疾病相关的4 000个基因的目标区域(包括全部外显子及上下游各30 bp内含子)进行杂交捕获,富集目的基因片段,使用 Illumina Nextseq500高通量测序仪进行基因测序(广州嘉检医学检测有限公司)。测序数据评估合格后通过bel2fastq软件进行生物信息学分析,与参考序列(Human GRCh 37/hg 19)进行匹配,生成变异列表。通过数据质量控制、变异频率和变异分类的筛选,以及变异与疾病的关系,锁定可能的致病突变。检测结果显示,患儿GTPBP 3基因存在杂合变异c.785 A>C(p.Q 262 p)、c.1169delG(p.G390Efs*16)杂合缺失F和c.1169delG(p.G390Efs*16)杂合缺失R(图2),分别来自患儿的母亲和父亲(均为杂合状态)。c.785 A>C(p.Q 262 p)导致第262 氨基酸由谷氨酰胺变异为脯氨酸,该变异尚未见文献报道,变异所在区域是这个蛋白质的一个核苷酸结合位点,不同物种的氨基酸序列高度保守,计算机辅助分析预测其可能影响蛋白质结构/功能,结合患儿临床表现和家系分析,依据美国ACMG变异分类指南(PMID:25741868,31690835),这个变异为“3类-意义未明”。c.1169delG(p.G390Efs*16)不仅导致15 个氨基酸被替换,而且还缺失120 个氨基酸,变异所在区域是这个蛋白的一个重要结构域,这个新型变异尚未见报道,该变异为移码变异,预测可能导致提前出现氨基酸的终止密码,结合患儿的临床表现和家系分析,依据ACMG指南(PMID:25741868,31690835),为“2类-可能致病”。

图2 患儿及父母基因测序图
2 讨论
线粒体普遍存在于真核细胞胞质中,是氧化和能量转换的重要场所,亦是细胞核外唯一带有遗传物质的细胞器。线粒体DNA(mtDNA)含有37个编码基因,分别编码22种tRNA、2种rRNA(12s和16srRNA)以及13种参与氧化磷酸化能量产生、呼吸链电子传递过程的蛋白亚单位[1]。呼吸链结构和功能缺损是构成人类线粒体疾病多样性的基础,既可由核DNA、也可由mtDNA 缺陷所致,可影响处于任何发育阶段的任何组织和系统,导致多样化的临床表现。线粒体病通常在婴儿期表现就很严重,这与氧化磷酸化障碍有关。tRNAs在线粒体翻译过程中具有非常重要的作用,其发挥正常功能需要进行转录后修饰,某些tRNAs 的修饰进化高度保守,不同物种中已发现不同的修饰侧链。在哺乳动物中,发现用于线粒体tRNAs修饰的是5-牛磺酸甲基尿苷[2]。而GTPBP3是一种高度保守的tRNAs 修饰酶,用于5-牛磺酸甲基尿苷的生物合成,tRNAs修饰有助于其提高稳定性、正确的折叠及解码能力[3]。GTPBP3基因变异患儿中,tRNAs更容易被降解。由于tRNAs结构变化,使其在蛋白质翻译过程中不能有效识别密码子,影响线粒体蛋白质翻译,最终导致线粒体呼吸链功能受损,能量供应不足,无氧酵解增加,导致乳酸性酸中毒;累及心脏,心肌供氧不足,导致心肌损害;活动时机体不能满足或代偿氧耗增加,导致急性呼吸衰竭;机体长期供氧不足,组织细胞代谢障碍,导致患儿生长发育迟缓和不同程度的脑损害。
关于GTPBP3基因变异导致线粒体翻译缺损相关疾病的临床研究报道罕见。有报道11例GTPBP3基因变异导致的与线粒体翻译缺损有关疾病的患儿,来自9个家庭。这些患儿有共同的临床表现,包括乳酸酸中毒(11/11)、心肌病(9/11)和神经系统相关症状(6/11),具体表现为喂养困难、生长发育迟缓、易疲劳、智力障碍、肌张力减低、视力损害和癫痫发作等。部分患儿头颅MRI提示丘脑、脑干、壳核、中脑、基底节等部位异常T 2 高信号;大部分患儿心脏彩超提示肥厚型心肌病或扩张型心肌病,这可能与心脏本身就是一个需氧量相对较高的器官,长期慢性缺氧心脏代偿性肥厚或者失代偿性扩张有关[3]。需要注意的是,疾病的严重性在不同的个体上差异明显,多数患儿在1 岁前就出现症状,有的从新生儿开始发病不久便死亡,有的从婴儿晚期开始出现症状,在青春期还存活。
GTPBP 3基因相关疾病为氧化磷酸化偶联缺陷23型,为常染色体隐性遗传。其特征是儿童早期出现肥厚型心肌病、心脏衰竭和/或神经系统症状,包括肌张力低下和精神运动发育延迟,某些可出现癫痫,实验室检查有与线粒体功能缺陷相关的乳酸性酸中毒。本例患儿的临床症状非常相似。本例患儿GTPBP 3基因的2个变异c.785A>C(p.Q262P)和c.1169delG(p.G390Efs*16)目前尚未在线粒体相关疾病中报道。GTPBP 3基因变异导致GTPBP 3 蛋白缺乏目前尚无特异的治疗方法,主要是对症支持治疗。患儿入院时呈卒中样发作综合征、神志昏迷、呼吸衰竭、动脉血pH 值低、乳酸高、心肌损害,经过气管插管、有创机械通气和静脉输注碳酸氢钠等治疗血气仍然不能改善,故入院后即给予“线粒体病鸡尾酒疗法”。目前常用此疗法来治疗线粒体脑病,包括使用核黄素、硫胺素、叶酸、肉毒碱、维生素C、维生素E和辅酶Q10等,使线粒体呼吸链的功能最大化,恢复线粒体的能量代谢。建议儿童可使用以下方案:核黄素100~400 mg/d,硫胺素50~100 mg/d,左旋肉碱50 mg/(kg·d),辅酶 Q1010~20 mg/(kg·d)[5 mg/(kg·d)]。此外,也有建议针对线粒体疾病高剂量补充亚叶酸0.5~8.0 mg/(kg·d)[4]。
连续性血液滤过和血液透析滤过是治疗代谢性危象的有效措施[5-6]。但是关于血液净化在儿科线粒体相关性代谢危象中应用的报道并不多。有报道采用持续性肾脏替代治疗25例代谢危象患儿,15例治疗成功、10例死亡;其中3例为线粒体相关性疾病,均表现为乳酸酸中毒,1例治疗成功,2例死亡[7]。本例患儿在机械通气和线粒体鸡尾酒疗法外,使用血液透析滤过数小时后动脉血气基本恢复正常。经治疗后患儿病情逐渐好转,顺利撤机,神志清醒。患儿出院后长期规律随访,目前病情稳定。
研究报道,GTPBP3基因敲除斑马鱼表现为心肌细胞肥大和心室肌纤维紊乱,产生肥厚性心肌病的表现[8]。有报道11例GTPBP 3基因变异导致与线粒体翻译缺损有关疾病的患儿中9例伴有心肌病。本例患儿超声心动图示左室稍增大,左室后壁动度降低,左心收缩功能降低,可能是GTPBP 3基因变异所致,当然也不除外严重代谢性酸中毒对心脏功能抑制所致。
综上,GTPBP3基因变异可影响患儿生长发育及全身多个系统。若发现患儿多系统受累,应高度怀疑遗传代谢性疾病,基因检测能明确诊断。当患儿出现难以纠正的乳酸酸中毒危及生命时,可采用血液透析以快速恢复正常的内环境稳态,以免对机体造成进一步的损害。
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国卒中杂志(2021年7期)2021-11-29
湖南饲料(2021年4期)2021-10-13
现代畜牧科技(2021年4期)2021-07-21
家庭医学·下半月(2019年5期)2019-07-12
腹腔镜外科杂志(2016年12期)2016-06-01
恋爱婚姻家庭·养生版(2016年2期)2016-02-17
药品评价(2015年11期)2015-12-08
