
结核分枝杆菌CYP143A1基因(Rv1785c)敲除菌株的构建
2021-10-18 01:27陈虹彤卢芸李国庆李聪然游雪甫杨信怡
中国医药生物技术 2021年5期
陈虹彤,卢芸,李国庆,李聪然,游雪甫,杨信怡
作者单位:100050 北京,中国医学科学院北京协和医学院医药生物技术研究所抗感染药物研究北京市重点实验室
细胞色素 P450(cytochrome P450,CYP)酶是一类含有血铁红素的单加氧酶超家族,广泛分布于各级生命体并发挥重要作用[1]。1998年,结核分枝杆菌(M.tuberculosis,Mtb)H37Rv 的全基因组序列图完成,研究者发现在全长 4.4 Mb 的MtbH37Rv 染色体 DNA 中存在 20 个不同的 CYP酶编码基因,其在基因组中的分布密度远高于多数其他原核微生物,这一现象提示 CYP 酶在Mtb中应该发挥着重要的生理功能[2-3]。随后的研究显示,CYP 酶参与Mtb呼吸链电子传递、脂质代谢与合成、胆固醇利用等诸多生理生化过程,对维持细菌生长增殖、细胞膜/壁完整、宿主细胞内持留、致病力、抗生素耐药性等颇为重要[4]。由于Mtb与真核生物间的 CYP 酶基因同源性较低(通常 <40%),且对应的电子传递体系亦存在显著差异,故被认为是一类可用于新型抗Mtb药物筛选、设计的潜在分子靶标[5-7]。然而,缘于其他物种与MtbCYP 编码基因总体同源性不高,以及Mtb自身 20 种CYP 酶编码基因之间序列同源性亦偏低,目前仅少数 CYP 亚型(如 CYP121A1、CYP51B1、CYP124A1、CYP125A1、CYP142A1)的结构、功能获得深入解析,多数亚型在Mtb中的作用和地位仍不甚明了[1,8]。例如,即便是系统进化树分析显示最为密切的同族酶 CYP135A1 与 CYP135B1之间也仅具 40% 的氨基酸同源性[7]。因此,要系统、客观了解 CYP 在Mtb形态、生存、毒力、环境适应等生命环节中所扮演的角色,以及有效推动 CYP 在抗Mtb药物可靶性方面的进展,对更多MtbCYP 的基因功能进行研究和探索具有重要意义[8-10]。
在细菌 CYP 酶介导的酶促反应中,铁氧还蛋白必不可少。这类蛋白通常含血铁红素辅因子,负责将相应铁氧还蛋白还原酶(FNR)从 NAD(P)H获得的电子传递至 CYP 酶(NAD(P)H → FNR →铁氧还蛋白 → CYP 酶),从而催化底物的代谢[11]。在MtbH37Rv 基因组中,仅发现有 5 个铁氧还蛋白和 2 个铁氧还蛋白-FNR 融合蛋白编码基因,远少于 CYP 酶编码基因数量。因此一个铁氧还蛋白作为电子供体蛋白,往往对应多个 CYP 酶,具有交通枢纽(Hub)的性质[12]。研究者在对 H37Rv和分枝杆菌属多个基因组序列进行对比分析时,发现相邻的 CYP143A1-铁氧还蛋白 Rv1786 基因对(Rv1785c-Rv1786)具有高保守性,提示两个蛋白可能属于少见的固定搭配,加之 CYP143A1 处于独立的进化树分支,均反映出该蛋白在分枝杆菌属可能介导独特而重要的功能,具有成为抗Mtb药物靶标的潜能[13]。另有研究显示Rv1785c属于esx-5位点的一部分,该位点的基因大多参与编码ESX-5 蛋白分泌系统的底物或组成部分[4]。而MtbESX-5 分泌系统(在Mtb中存在 5 类 VII 型分泌系统,分别命名为 ESX-1~ESX-5[14])通常参与诸如抑制巨噬细胞促炎细胞因子分泌、诱导caspase 依赖性细胞死亡等重要生理学功能[15-16]。鉴于Rv1785c与esx-5位点的上述潜在关联性,推测 CYP143A1 也与Mtb的感染、侵袭功能有关。因此,深入探究 CYP143A1 基因的生理功能具有重要价值。
研究MtbCYP143A1 基因生理功能,首要任务是构建该基因的敲除菌株。本研究采用噬菌体介导的基因敲除技术[17],通过构建同源交换位点、构建穿梭质粒、构建分枝杆菌噬菌体并转染MtbH37Rv,成功构建了MtbH37RvRv1785c敲除菌株,为后续进一步探索 CYP143A1 基因功能奠定了基础。
1 材料与方法
1.1 材料
1.1.2 试剂与培养基 胰蛋白胨、酵母提取物、琼脂粉、LB Broth、Middlebrook 7H9、Middlebrook 7H10 及 OADC 均购自美国 BD 公司;氯化钠、甘油购自生工生物工程(上海)股份有限公司;潮霉素B(hygromycin B,HYG)、吐温 80 购自美国Sigma 公司;胶回收试剂盒 EasyPure®Quick Gel Extraction Kit 、 Trans2KPlusII DNA Marker 、Trans15K DNA Marker 购自全式金生物技术有限公司;T4 DNA 连接酶、限制性内切酶PacI 购自宝生物工程(大连)有限公司;Fast DigestVan91I、Phusion High Fidelity DNA Polymerase 购自美国Thermo Fisher 公司;质粒提取试剂盒购自天根生化科技(北京)有限公司;细菌基因组提取试剂盒购自美国 Omega 公司;包装试剂盒购自 Epicentre生物技术公司。
顶层琼脂(100 ml):Middlebrook 7H9 broth 0.47 g、琼脂 0.75 g,121 ℃ 高温灭菌 15 min;MP 缓冲液:50 mmol/L Tris-HCl、150 mmol/L NaCl、10 mmol/L MgSO4、2 mmol/L CaCl2,稀盐酸调至 pH 7.5 或 7.8,0.22 μm 无菌滤器过滤。
1.1.3 细菌培养E.coliDH5α、E.coliHB101采用 LB 液体培养基或 LB 固体培养基培养;M.smegmatismc2155、MtbH37Rv 采用 7H9 液体培养基或 7H10 固体培养基培养。
1.2 方法
1.2.1 构建 p0004s-△Rv1785c质粒 以MtbH37Rv DNA 为模板,根据待敲除基因Rv1785c上下游序列基因片段(左臂和右臂),设计引物对LFP/LRP(左臂上游引物/左臂下游引物)和RFP/RRP(右臂上游引物/右臂下游引物),其中引物 LRP 和 RFP 分别包含Rv1785c左、右端部分DNA 序列,引物序列见表 1。采用引物对 LFP/LRP、RFP/RRP,Thermo Scientific 公司 Phusion 高保真DNA 聚合酶进行 PCR,分别扩增Rv1785c左臂(720 bp)、右臂(608 bp)DNA 序列。PCR 扩增反应体系选用 GC buffer 并额外添加 8% DMSO。反应条件:预变性 98 ℃,30 s;变性 98 ℃,10 s,退火 55 ℃,30 s,延伸72 ℃,30 s/kb,30 个循环;延伸 72 ℃,5 min。
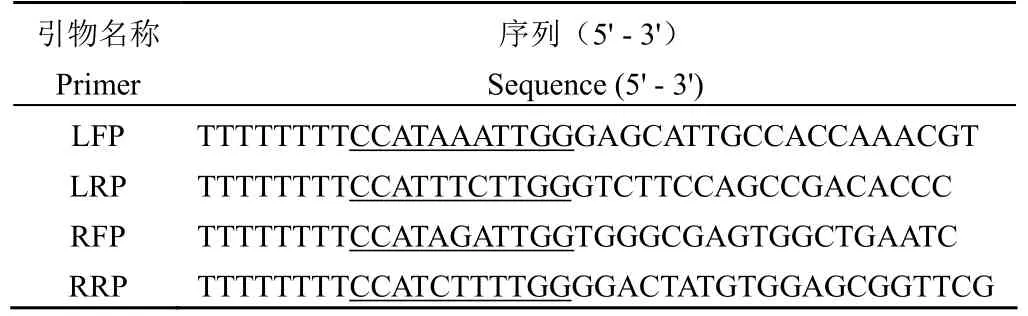
表1 左、右臂上下游引物序列Table 1 Upstream and downstream primers of left and right arm
采用琼脂糖核酸凝胶电泳对 PCR 扩增产物进行验证,使用胶回收纯化试剂盒回收目的 DNA 片段。随后使用质粒提取试剂盒提取 p0004s 质粒,用Van91I 限制性内切酶分别酶切左、右臂 DNA片段及 p0004s 质粒后,使用快速回收试剂盒分别回收酶切后的目的 DNA 片段。
将获得的左臂、右臂及 p0004s 质粒酶切片段采用 T4 DNA ligase 连接,转化E.coliDH5α 感受态细胞,涂布于含有 150 μg/ml HYG 的 LB 琼脂平板。筛选阳性克隆子,通过测序进行质粒验证,将阳性质粒保存备用。
1.2.2 构建 phAE159-△Rv1785c噬菌粒 采用质粒提取试剂盒提取 phAE159 质粒以及 p0004s-△Rv1785c阳性质粒,分别使用限制性内切酶PacI酶切质粒并回收目的片段。用 T4 DNA ligase 将两个酶切后的线性质粒片段进行连接,连接产物用试剂盒辅助转化E.coliHB101 感受态细胞,涂布于含 150 μg/ml HYG 的 LB 平板。挑取该抗性平板上生长的单菌落至含 150 μg/ml HYG 的 LB 液体培养基中,37 ℃ 振荡培养。抽提噬菌粒,使用PacI限制性内切酶酶切,并用琼脂糖凝胶电泳验证。
1.2.3 制备噬菌体
在例(5)和(6)中时间词或空间词被省略或被隐含在句子之中不被指出,这些成分可以补充,但没有补充的必要。例(5)中茶果端上来自然是要放在桌几之上的,此时处所词未曾出现,但隐含在以上句子之中;同理例(6)之中,这位年轻的公子按常理必然是从房门而入,众人皆知没有提及的必要,在这个句子之中,存在主体-年轻的公子是突出重点,其他次要元素可以忽略不计。
1.2.3.1 制备及培养M.smegmatismc2155 电转感受态细胞 将新鲜M.smegmatismc2155 单菌落接种至 5 ml 7H9 液体培养基,37 ℃ 振荡培养至对数生长期(OD600nm=0.5~1.0,1~2 d);将培养液按 1:100 比例接种于 100 ml 7H9 培养基,37 ℃过夜培养至OD600nm≈ 0.6;将培养物在冰上放置0.5~1 h 后,4 ℃、5000 r/min 条件下离心 10 min;收集菌体并用预冷 10% 无菌甘油洗涤菌体 2 次,加入 10 ml 预冷 10% 无菌甘油,充分混匀后分装并于–80 ℃ 冻存备用。
1.2.3.2 电转 取 5~10 μl 阳性 phAE159-△Rv1785c噬菌粒与 200 μlM.smegmatismc2155感受态细胞混匀,使用 2 mm 电转杯 Bio-Rad 电转仪电击转化(电击参数:电压 2.5 kV,电阻 1000 Ω,电容 25 μF)。电击后,加入 1 ml 7H9 培养基,37 ℃ 过夜培养复苏。将菌液与 4 ml 顶层琼脂混匀,铺于 7H10 平板,30 ℃ 培养 2~3 d,观察是否形成噬菌斑(空斑)。
1.2.3.3 扩增高滴度噬菌体 从平板上挑取4~5 个含有噬菌体的空斑,加入 200 μl MP 缓冲液,4 ℃ 孵育过夜。将其与 300 μl 新鲜培养M.smegmatismc2155 菌液混合后,再与适量顶层琼脂混匀并铺板。30 ℃ 培养 2~3 d 后,在含有噬菌斑的平板上加入适量 MP 缓冲液,4 ℃ 放置过夜,采用 0.22 μm 无菌滤器过滤上清液以收集高滴度噬菌体,于 4 ℃ 保存。
1.2.4 构建 H37RvRv1785c敲除菌株 将培养至对数生长期的 H37Rv 预先采用 MP 缓冲液洗涤,按感染复数(MOI)为细菌:噬菌体=1:10 取高滴度噬菌体感染 H37Rv,37 ℃ 孵育过夜。离心后弃上清,加入适量新鲜 7H9 液体培养基,37 ℃孵育过夜,离心后弃上清。收集菌体,涂布于含75 μg/ml HYG 的 7H10 + OADC 平板,37 ℃ 培养4~5 周。挑取单克隆接种至含 75 μg/ml HYG 的7H9 + OADC 液体培养基,37 ℃ 培养 4~5 周后,提取基因组,进行基因测序并通过 PCR 验证敲除结果。
1.2.5 PCR 验证 分别设计左、右臂上下游引物LYZFP/LYZRP 和 RYZFP/RYZRP,引物序列见表 2。在 LFP 引物匹配序列上游 100~200 bp 处设计上游验证引物 LYZFP,在 RRP 引物匹配序列下游 100~200 bp 处设计下游验证引物 RYZRP。LYZRP 引物设计于sacB基因,RYZFP 引物设计于hyg基因。
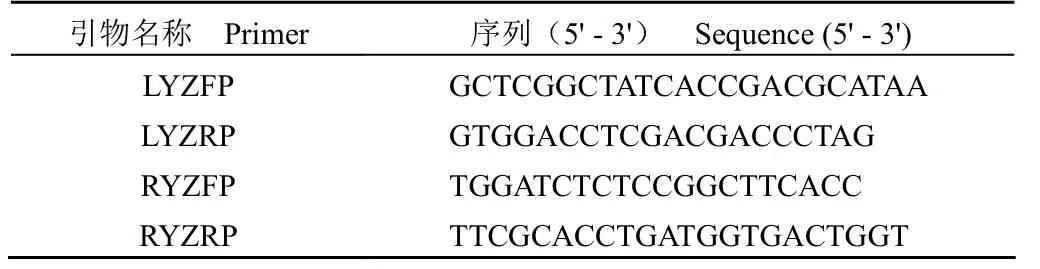
表2 敲除验证引物序列Table 2 Primers used in PCR verification
1.2.5.1 两片段法 PCR 验证基因敲除菌株 两片段法 PCR 验证基因敲除菌株原理如图 1。分别使用引物对 LYZFP/LYZRP 和 RYZFP/RYZRP,以敲除菌株基因组(MUT strain)为模板扩增 DNA 片段,以野生型菌株基因组(WT strain)作为对照组模板进行 DNA 扩增。PCR 产物进行琼脂糖凝胶电泳检测,验证目的基因敲除结果。
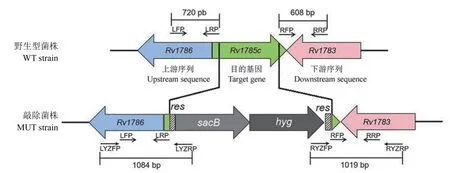
图1 两片段法 PCR 验证原理Figure 1 Principle of two-segment PCR
1.2.5.2 全长法 PCR 验证基因敲除菌株 全长法 PCR 验证目的基因敲除菌株原理如图 2。使用引物对 LYZFP/RYZRP,以敲除菌株基因组为模板进行 DNA 扩增,以野生型菌株基因组模板作为对照组进行 DNA 扩增。PCR 产物进行琼脂糖凝胶电泳检测,验证目的基因敲除结果。
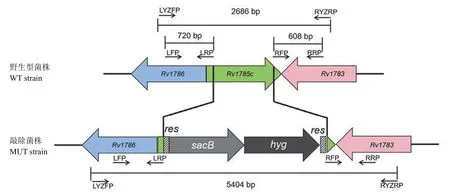
图2 全长法 PCR 验证原理Figure 2 Principle of full-length PCR
2 结果
2.1 PCR 扩增左右臂
分别使用引物对 LFP/LRP 和 RFP/RRP,PCR扩增Rv1785c基因左臂和右臂 DNA 片段,采用琼脂糖凝胶电泳验证扩增结果,电泳结果见图 3。扩增得到了大小分别约为 720 bp 及 608 bp 的DNA 片段,与预期目的条带一致。PCR 电泳结果表明Rv1785c左、右臂 DNA 扩增成功。
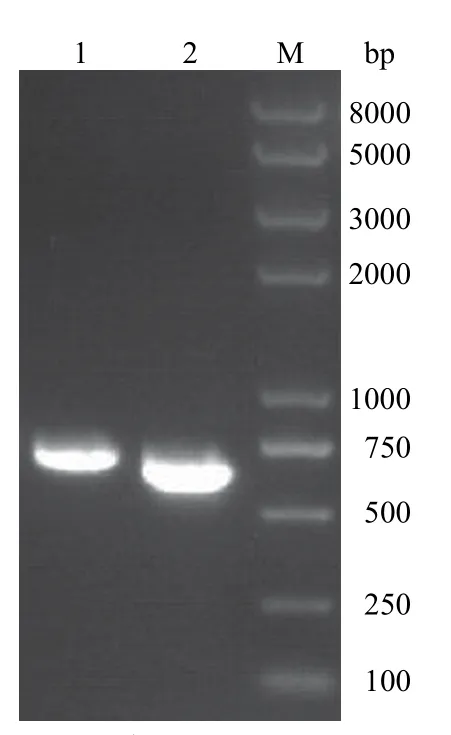
图3 Rv1785c 基因左、右臂 DNA 扩增核酸电泳Figure 3 Electrophoresis of homology arms of Rv1785c
连接左右臂及 p0004s 质粒,转化E.coliDH5α感受态细胞并涂板。对获得的阳性克隆子抽提质粒并测序验证,测序结果显示 p0004s-△Rv1785c质粒构建成功。
2.2 phAE159-△Rv1785c 噬菌粒的构建
利用PacI 酶切 p0004s-△Rv1785c质粒及phAE159 质粒并回收,连接片段、包装并转化E.coliHB101,筛选获得的阳性噬菌粒。挑取 3 个单克隆菌落,分别抽提噬菌粒,酶切后进行核酸凝胶电泳验证,电泳结果见图 4,获得片段大小分别约为 7 kb 及 >10 kb 的条带,与预期 p0004s-△Rv1785c质粒及 phAE159 质粒条带大小一致,结果表明 phAE159-△Rv1785c噬菌粒构建成功。

图4 phAE159-△Rv1785c 噬菌粒酶切验证电泳Figure 4 Electrophoresis of enzyme-digested product of phAE159-△Rv1785c
2.3 噬菌体的制备
取适量构建成功的噬菌粒DNA电转化M.smegmatismc2155 感受态细胞,铺板,30 ℃ 培养 2~3 d 后观察是否形成噬菌体空斑。平板培养结果如图5所示,可观察到平板上有空斑均匀、随机分布。

图5 噬菌体空斑Figure 5 Plaque of phage
2.4 Mtb H37Rv CYP143A1 敲除株的构建及验证
制备高滴度噬菌体,感染MtbH37Rv 并涂布于含 75 μg/ml HYG 的 7H10 + OADC 平板,37 ℃培养 4~5 周后,平板上有Mtb单克隆菌落生长。挑取单克隆接种至含 75 μg/ml HYG 的 7H9 +OADC 液体培养基中培养,提取基因组,分别采用两片段法及全长法 PCR 及基因测序验证基因敲除结果。
2.4.1 两片段法 PCR 验证基因敲除菌株 根据两片段法 PCR 验证原理,以Rv1785c敲除菌株基因组为模板,分别使用引物对 LYZFP/LYZRP 和RYZFP/RYZRP,扩增可得到大小为 1084 bp 及1019 bp 的条带。电泳结果见图 6,以敲除株基因组为模板,分别使用引物对 LYZFP/LYZRP 和RYZFP/RYZRP,均扩增出大小在 1 kb 左右的DNA 片段,与预期目的条带大小一致;当以野生型菌株基因组为模板进行扩增时,则无法扩增出目的 DNA 片段。两片段法 PCR 验证结果表明目的基因Rv1785c敲除成功。
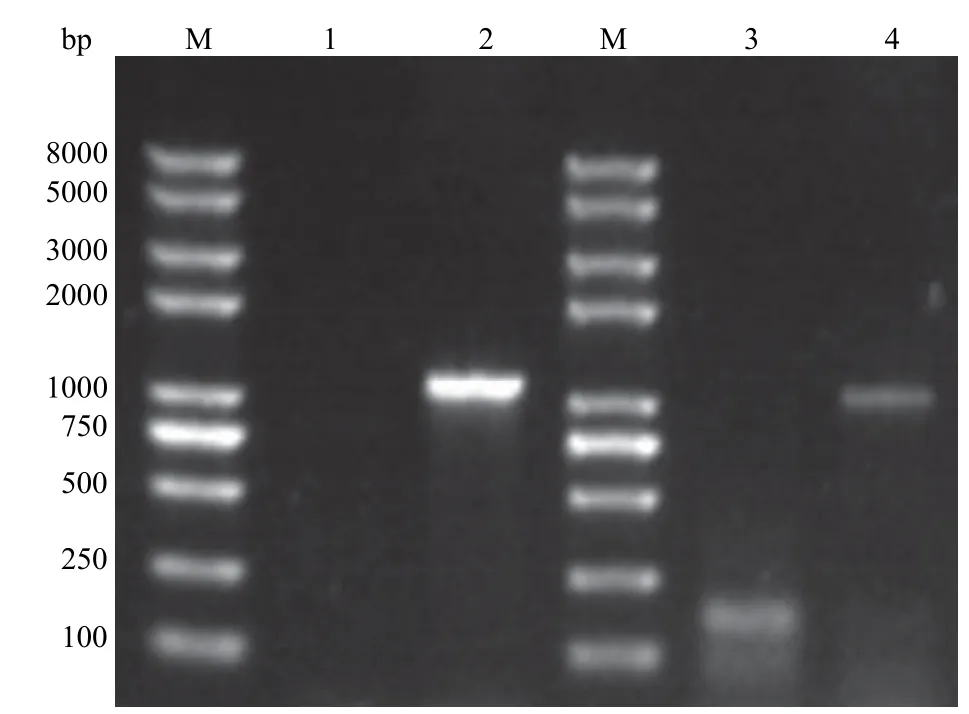
图6 两片段法 PCR 验证基因敲除结果Figure 6 Result of two-segment PCR
2.4.2 全长法 PCR 验证基因敲除菌株 根据全长法 PCR 验证原理,以野生株基因组为模板,扩增应得到包含左、右臂及完整Rv1785c全长2686 bp 的条带,而敲除株由于Rv1785c被sacB及hyg区域置换,扩增会得到大小为 5404 bp 的条带。电泳结果见图 7,使用引物对 LYZFP/RYZRP并以敲除菌株基因组为模板扩增时获得了约 5.5 kb大小的 DNA 片段,而以野生型菌株基因组模板为平行对照,PCR 扩增获得大小约 2.7 kb 的 DNA片段,与预期结果一致。全长法 PCR 验证结果显示,Rv1785c已被sacB+hyg区域替换,H37Rv CYP143A1 基因敲除菌株构建成功。

图7 全长法 PCR 验证基因敲除结果Figure 7 Result of full-length PCR
2.4.3 敲除株基因测序 序列测定结果如图 8所示,与 PCR 验证结果一致,目的基因Rv1785c已被sacB+hyg B区域替换,MtbH37RvRv1785c敲除菌株构建成功。
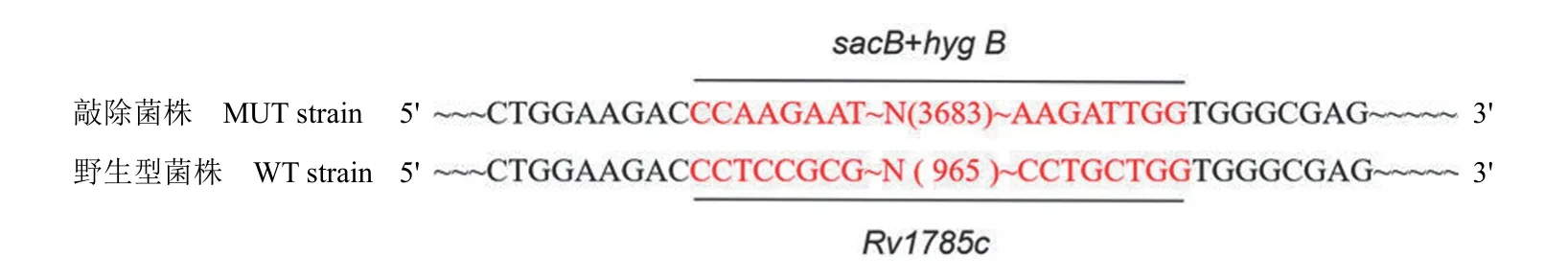
图8 敲除菌株基因测序Figure 8 Gene sequencing result of Rv1785c knockout strain
3 讨论
结核病(TB)是一种古老的传染病,是由Mtb感染引起的。根据 WHO 新发布《全球结核病报告》,2019年全球有近 1000 万 TB 新发病例[18]。然而,一方面由于Mtb与 HIV 合并感染更具难治性[19-20],另一方面由于Mtb对一线抗 TB 药物乙胺丁醇、异烟肼、利福平等日益耐药,尤其随着耐多药(MDR)、泛耐药(XDR)、完全耐药(TDR)菌株的广泛传播,对全球 TB 的治疗管控提出了重大挑战[18]。近几年虽逐渐有抗结核新药上市,如FDA 批准的新药贝达喹啉及德拉马尼,但相对于日益严峻的Mtb耐药问题,开发中的抗 TB 药物无论品种还是数量仍然远远不够。其中,显著的短板是抗结核药物新型靶标的发现和利用仍不尽人意。
利用Mtb基因结构与功能的研究,有助于识别和确认新的抗 TB 药物靶点。通过基因敲除技术,构建该基因位点的空等位基因菌株,是表征基因功能的一个有效且常用的手段。然而,由于Mtb的细胞壁较一般细菌更厚,使得质粒等遗传工具较难进入细胞内;Mtb生长缓慢,生长周期长,敲除株构建实验周期较长;且Mtb基因组同源重组率较低,使得Mtb敲除株的构建有较大的难度[21];目前有效的基因工程技术较为缺乏,常用的CRISPR-Cas9 基因敲除技术尚未能成功应用于敲除Mtb基因,仍需深入研究其在Mtb中发挥功能的机制。
本实验采用含温敏型噬菌体元件的穿梭质粒phAE159,相对于另一常用质粒 phAE87,phAE159具有更高的克隆能力和 DNA 传递率。其在 30 ℃环境下以噬菌体形式进行复制,但在 37 ℃ 时不进行复制,而是在分枝杆菌内注入 DNA[22]。phAE159可容纳大片段 DNA,可将长达 10 kb 的重组DNA 传递至分枝杆菌胞内[17]。因 phAE159 含有分枝杆菌噬菌体编码元件,结合重组与噬菌体转导的方法,构建重组 phAE159-△Rv1785c噬菌粒,在M.smegmatismc2155 中进行噬菌体扩增,并侵染MtbH37Rv 感受态细胞,可有效获得MtbH37Rv 目标突变体,实现高效同源重组。采用本法,前期实验在E.coli和M.smegmatis中进行,使实验周期显著缩短[22]。此法的主要不足是步骤相对较繁琐,需构建同源臂、包装噬菌体、感染菌株等步骤来实现敲除株的构建。
总之,本研究构建了包含Rv1785c上下游同源臂(左臂及右臂)的等位交换质粒,并将该质粒与含有分枝杆菌温敏型噬菌体元件的穿梭质粒phAE159 连接获得重组噬菌粒,最终经转导进入MtbH37Rv,成功构建MtbH37Rv CYP143A1 基因敲除菌株,为后续开展该基因功能、可靶性的研究及该基因与esx-5位点的潜在关联性研究奠定了重要物质基础。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
昆明医科大学学报(2022年2期)2022-03-29
植物保护(2021年4期)2021-11-12
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
科学24小时(2020年4期)2020-05-14
青年文学家(2017年28期)2017-11-28
中国新闻周刊(2017年19期)2017-06-01
少林与太极(2014年9期)2014-10-15
