
高效液相色谱-串联质谱法定量测定大鼠血浆中LMV-12(HE003)及其代谢产物M4
2021-10-18 01:27刘淑洁闻镍王宇黄舒佳淡墨汤瑶王晓霞陶琳耿兴超王三龙刘丽
中国医药生物技术 2021年5期
刘淑洁,闻镍,王宇,黄舒佳,淡墨,汤瑶,王晓霞,陶琳,耿兴超,王三龙,刘丽
作者单位:100176 北京,中国食品药品检定研究院国家药物安全评价监测中心药物非临床安全评价研究北京市重点实验室(刘淑洁、闻镍、王宇、黄舒佳、淡墨、汤瑶、王晓霞、耿兴超、王三龙、刘丽);100022北京,国家药品监督管理局药品审评中心(刘淑洁);330096 南昌,南昌弘益药业有限公司(陶琳)
LMV-12(HE003)(以下简称 LMV-12)为小分子酪氨酸激酶抑制剂,靶向抑制 c-Met、VEGFR2及 RET 等信号通路而发挥抗肿瘤作用,杀死肿瘤细胞,减少转移并抑制血管生成。本品是我国自主研发的创新型小分子抗肿瘤药物,前期研究发现该化合物对人肺癌细胞和人胃癌细胞裸鼠皮下移植瘤均有明显的生长抑制作用,但该化合物在动物体内的安全性尚待研究。同时前期药代研究发现该化合物的代谢产物 M4 在体内暴露量较高,且与其抑制肿瘤的作用机制密切相关,故该代谢产物的安全性和体内暴露也值得关注[1-3]。在非临床安全性研究中开展伴随的毒代动力学研究能够评价药物暴露与毒性反应的关系,可靠的生物分析方法是开展毒代动力学研究的前提和基础[1-3]。LMV-12 及其代谢产物在大鼠体内血药浓度测定的生物分析方法及方法学验证尚未开展,亟需建立有效可靠的分析方法、开展验证以支持相关临床前和临床研究。
本研究优化样品前处理和检测条件,采用HPLC-MS/MS 方法,建立了同时实现 SD 大鼠血浆内 LMV-12 及代谢产物 M4 的定量研究方法,并开展了完整的方法学验证,证明该方法的选择性、残留、标准曲线、定量下限、准确度与精密度、基质效应、稀释可靠性等符合要求,并对生物基质中的待测物稳定性进行了考察。为 LMV-12 的药代动力学和毒代动力学研究提供了可靠的生物分析方法。
1 材料与方法
1.1 材料
1.1.1 受试物和内标 LMV-12(HE003),白色粉末,无臭,纯度 99.6%;M4,类白色粉末,纯度98.2%;CH3-LMV-12,白色粉末,纯度 99.2%,均由南昌弘益药业有限公司提供。
1.1.2 试剂 乙腈(HPLC 纯)、甲醇(HPLC 纯)、甲酸(LC/MS 纯)均为美国 Fisher scientific 公司产品;甲酸铵(质谱纯),Fluka;DMSO(GC 纯)购自美国 Sigma 公司;超纯水由 Millipore Milli-Q Advantage A10 超纯水机当日制备。
生物基质:SD 大鼠混合血浆和 SD 大鼠空白个体血浆均自行制备,–70 ℃ 冻存。SD 大鼠混合血浆由 6 只以上 SD 大鼠空白个体血浆混合而得。
1.1.3 仪器 HPLC-MS/MS 仪(Accela 高效液相色谱仪、TSQ Quantum Access 三重四级杆串联质谱仪、电喷雾离子源(ESI)、Xcalibur 2.2 工作站)均购自美国 Thermo Fisher 公司;5415R 离心机购自 Eppendorf 公司;Milli-Q Advantage A10 型号超纯水机购自 Millipore 公司;AX 205 和 PB 203-N型号电子天平购自 Mettler Toledo 公司;Vortex G560E 涡旋仪购自 Scientific Industries 公司。
1.2 方法
1.2.1 标准储备液及标准工作液的配制 分别精密称取适量 LMV-12 对照品、M4 对照品置于量瓶中,加稀释液(50% 乙腈-水)使溶解并稀释至刻度,摇匀,得到浓度为 0.2 mg/ml 的 LMV-12 对照品储备液和 M4 对照品储备液。储备液用稀释液系列稀释后,得到系列标准工作液。
1.2.2 内标储备液及工作液的配制 精密称取CH3-LMV-12 对照品 10 mg,置 50 ml 量瓶中,加200 μl DMSO 溶解并用稀释液稀释至刻度,摇匀,得到浓度为 0.2 mg/ml 的内标储备液,进而用0.05% 甲酸-乙腈稀释得到内标工作液(20 ng/ml)。1.2.3 质控储备液及工作液的配制 质控储备液独立配制,配制方法同标准储备液。用稀释液系列稀释后,得到目标浓度的质控样品。
1.2.4 高效液相色谱条件 采用 ACQUITY HPLC®CSH C18 色谱柱(2.1 mm × 50 mm,1.7 µm),配有保护柱 Phenomenex C18(4.0 mm × 3.0 mm);流动相:A 相:乙腈,B 相:0.3% 甲酸-40 mmol/L甲酸铵水溶液,梯度洗脱条件如表 1,柱温 40 ℃;进样量 3 μl。

表1 洗脱梯度设置表Table 1 Elution gradient settings
1.2.5 质谱条件 采用电喷雾离子化电离源(ESI),喷雾电压 3500 V;加热毛细管温度 350 ℃;鞘气氮气,流速 45 Arb;辅助气氮气,流速 15 Arb;碰撞气氩气,流速 1.5 mTorr。LMV-12、M4 和内标的二级碰撞能量分别为 32、30 和 29 eV;正离子方式检测;扫描方式为选择反应监测(SRM),用于定量分析的离子反应为m/z674.300 → 169.200(LMV-12),m/z661.200 → 156.000(M4)和m/z688.200 → 183.000(CH3-LMV-12);扫描时间为0.5 s。
1.2.6 标准样品和质控(QC)样品的制备 分别精密量取 SD 大鼠空白混合血浆,添加 LMV-12和 M4 系列标准工作液,得到 LMV-12 浓度为 20、50、100、200、500、1000、2000、5000、8000 ng/ml以及 M4 浓度为 5、12.5、25、50、125、250、500、1250、2000 ng/ml 的校正标样。
质控样品配制方法同标准样品。质控样品中LMV-12 浓度分别为 20、60、600、6000 ng/ml,M4 浓度分别为 5、15、150、1500 ng/ml。
1.2.7 血浆样品前处理方法 采用蛋白沉淀方法[4-5]去除血浆中的蛋白,同时提取待测物质:准确吸取血浆样本 50 μl,加入内标工作液 200 μl,涡旋 2 min 后,12 000 r/min、4 ℃ 离心 10 min,吸取上清液,转移至进样衬管中,进样 3 μl 测定。
另取 SD 大鼠空白混合血浆(不添加任何物质)加入 0.05% 甲酸-乙腈 200 μl,涡旋 2 min 后,12 000 r/min、4 ℃ 离心 10 min,吸取上清液,作为空白样品。
1.2.8 分析方法验证项目及标准 按照《中国药典》现行版《生物样品定量分析方法验证指导原则》[6]以及《化学药物非临床药代动力学研究技术指导原则》[1]相关要求,参考 FDA《药品和生物制品的分析程序和方法验证行业指南》[7]、EMA《生物样品分析方法验证指导原则》[8]以及 ICH《M10:生物样品分析方法验证》[9],开展完整方法学验证,考察该方法的选择性、残留、标准曲线、准确度和精密度、基质效应、回收率、定量下限和稀释可靠性,并考察样品保存稳定性。
研究中±s用 Excel 进行计算,计算公式如下,判定标准按照现行版《生物样品定量分析方法验证指导原则》[6]执行。
准确度:准确度用测定值与理论值的偏离程度,即偏差(Deviation,Diff%)表示;计算公式如下:Diff%=(测得浓度–标示浓度)/标示浓度 ×100%
精密度:精密度用多次测定数据的相对标准偏差(relative standard deviation,RSD%)表示。
RSD%=测得浓度 SD 值/测得浓度均值 ×100%
1.2.9 SD 大鼠毒代动力学研究 SD 大鼠雌雄各 8 只,经口灌胃给予 LMV-12 受试物 28 d,每天一次,给药剂量 15 mg/kg。于首次和末次给药前、给药后 0.5、1、2、4、6、8、24 h,共 8 个采血点采血。采集血液置于 EDTA 抗凝离心管中4000 r/min,4 ℃,10 min 离心后,收集血浆分装2 管,其中 1 份保证在 200 μl 以上;剩余的全部置于另 1 个收集管中,置于–65 ℃ 以下条件保存待测。
2 结果
2.1 质谱条件的优化
由于 LMV-12 尚无同位素内标,故选取结构相似的 CH3-LMV-12 为内标。取 LMV-12、M4 和内标三种化合物的储备液,分别稀释至 0.1 mg/ml注入 ESI 离子源中,用正离子模式得到各自稳定的准分子离子峰与特征碎片离子。然后将 HPLC与串联四极杆质谱仪连接,选择各自的监测离子对,分别对离子传输管温度、透镜电压、鞘气压、辅助气压等进行优化,使样品的离子化效率达到最佳。最终确定了质谱条件。用于定量分析的离子反应为m/z674.300 → 169.200(LMV-12),m/z661.200 → 156.000(M4)和m/z688.200 → 183.000(内标)。
2.2 色谱条件的优化
良好的色谱条件对定量结果有重要意义。前期方法开发中发现 LMV-12 化合物表现极为特殊,其残留严重,可达中浓度质控(MQC)水平,经探索发现其对所处溶液环境的极性极为敏感,本研究从液相条件如色谱柱的选择、流动相、洗脱程序设计等方面分别优化。
经筛选,选用 Waters ACQUITY HPLC®CSH C18 色谱柱(2.1 mm × 50 mm,1.7 µm)。该色谱柱为新一代杂化颗粒的 HPLC 超高压液相色谱柱,CSH 颗粒在桥式亚乙基杂化颗粒技术的基础上在其表面控制少量电荷,这种修饰能够改善离子化小分子的峰形、载量行为和峰容量,并且使得在LMV-12 的分离中表现出较低的残留。
在流动相中的水相中适当添加甲酸及甲酸铵以提高待测物离子化效率,同时应避免流动相中离子浓度过高导致离子抑制,经过反复优化最终采用0.3% 甲酸-40 mmol/L 甲酸铵水溶液和乙腈为流动相。
洗脱程序中较大梯度变化将导致溶剂极性环境变化大而造成 LMV-12 的高残留,采用 40%~60% 的弱梯度、较缓变化的梯度,可同时实现溶液中基质内源性物质以及多种化合物的有效分离,同时有效控制 LMV-12、M4 和内标三种化合物的残留在指导原则规定的 20% 定量下限以下。
通常,较高的流速有利于尖锐和对称的峰形,但本研究发现 LMV-12 在较高的流速下会因溶液极性变化快而出现高残留,适当降低流速有利于降低残留,经优化最终采用流速为 250 μl/min。最终梯度洗脱程序如表 1所示。
在最终色谱条件下 LMV-12 保留时间为1.13 min,M4 的保留时间约为 1.22 min,内标的保留时间约为 1.40 min,三种化合物均可实现较对称色谱峰,峰形较窄且灵敏度高,同时残留符合要求。
2.3 选择性
采用来自 6 只个体 SD 大鼠的空白血浆样品考察基质内源性物质对待测物及内标物的干扰。上述个体血浆经前处理后,按照已建立的分离检测条件分析,空白基质在 LMV-12 保留时间处的响应不超过 1.55%,M4 保留时间处的响应不超过1.30%,内标处的响应不超过 0.01%(图 1)。另外,分别单独进样 LMV-12、M4 和内标,结果显示每种化合物对其他两种组分的影响不超过 8.25%。本方法内源性组分对待测物干扰以及三种化合物之间的相互干扰均满足《生物样品定量分析方法验证指导原则》的要求[6-9]。

图1 空白血浆中 LMV-12、M4 和内标的图谱(A)和定量下限血浆样品中 LMV-12、M4 和内标的图谱(B)Figure 1 Spectrum of LMV-12,M4 and internal standard in blank plasma (A) and in LLOQ samples (B)
2.4 残留
在注射定量上限(upper limit of quantification,ULOQ)标样后,注射空白样品,其中 LMV-12 的峰面积不大于注射定量下限(lower limit of quantification,LLOQ)中 LMV-12 峰面积的 0.13%,M4 的峰面积不大于 LLOQ 中 M4 峰面积的7.10%,内标峰面积不大于 LLOQ 内标峰面积的0.01%,满足指导原则对于干扰组分不高于待测物LLOQ 峰面积 20%、内标峰面积 5% 要求[6-9]。
2.5 基质效应
6 批来自不同个体的 SD 大鼠空白血浆(其中含 2 个溶血血浆)提取后,分别加入低、高质控两个浓度水平的 LMV-12、M4 及内标,分别与同浓度水平不含血浆基质的 LMV-12、M4 及内标纯溶液的相应峰面积相比,绝对基质因子分别为0.98、0.93、1.08,LMV-12 和 M4 归一化的基质因子分别为 0.92、0.86,RSD 分别为 9.40% 和12.98%,符合指导原则中 RSD 不大于 15% 的标准。本品不涉及影响脂质代谢,故未在临床前研究中考察高脂基质的基质效应,建议后续临床研究补充开展高脂基质的基质效应考察[7]。
2.6 标准曲线和灵敏度
以待测物与内标物峰面积比值为纵坐标(y),以待测物的理论浓度为横坐标(x),采用线性最小二乘法(权重系数 1/x2)求得标准曲线,LMV-12和 M4 各评价 4 条标准曲线,所有标准曲线线性相关性良好(r2>0.99),符合接受标准。LMV-12 浓度范围为 20~8000 ng/ml,M4 浓度范围为 5~1500 ng/ml,代表性标准曲线拟合图见图 2。两种待测物定量范围可以大致覆盖预期的大鼠毒性研究药物血浆暴露浓度,描述 LMV-12 和 M4 的毒代动力学特征,灵敏度基本满足测定需求。

图2 血浆中 LMV-12(A)和 M4(B)定量分析标准曲线图Figure 2 Calibration curve for quantitative analysis of LMV-12 (A) and M4 (B) in plasma
2.7 定量下限与准确度、精密度
定量下限及低、中、高水平的质控样品(LMV-12 浓度分别为 20、60、600、6000 ng/ml,M4 浓度分别为 5、15、150、1500 ng/ml),每个添加水平重复测定 6 个样品,连续测定 4 个分析批(3日内),以实测值与理论值之比计算方法的准确度,并计算批内与批间精密度(以 RSD% 计),具体结果见表 2。

表2 SD 大鼠血浆基质中的 LMV-12/M4 测定的准确度和精密度Table 2 Accuracy and precision of LMV-12/M4 in SD rat plasma matrix
经验证,LLOQ 样品中 LMV-12 的批内准确度偏差在–11.45%~9.06% 之间,精密度 RSD 不超过 17.86%;批间准确度偏差为 0.02%,批间精密度 RSD 为 8.53%。LLOQ 样品中的 M4 的批内准确度偏差在–10.95%~3.87% 之间,精密度RSD 不超过 15.65%;批间准确度偏差为–3.19%,批间精密度 RSD 为 7.39%。LMV-12 的定量下限为 20 ng/ml,M4 的定量下限为 5 ng/ml。
低、中、高浓度质控样品中 LMV-12 的批内准确度偏差在–9.4%~6.19% 之间,精密度 RSD不超过 9.70%;批间准确度偏差在–5.44%~–0.01%之间,批间精密度 RSD 不超过 4.73%。低、中、高浓度质控样品中 M4 的批内准确度偏差在–12.18%~3.80% 之间,精密度 RSD 不超过8.69%;批间准确度偏差在–8.57%~–1.93% 之间,批间精密度 RSD 不超过 4.95%。上述结果符合指导原则对生物样品测定分析方法验证的要求[6-9]。
2.8 提取回收率
将低、中、高 3 个质控水平的血浆样品(带内标),与基质提取液(带内标)中相应浓度待测物的峰面积相比,计算得到提取回收率,3 个批次 3 个浓度质控样品中 LMV-12 的回收率在85.89%~88.77% 之间,各浓度回收率的相对标准偏差(RSD)不高于 5.82%。M4 的回收率在88.38%~93.24% 之间,各浓度回收率的 RSD 不高于 8.87%。内标的平均回收率为 109.72%,RSD为 5.31%。
2.9 稀释可靠性
含 LMV-12 浓度 12 000 ng/ml、M4 浓度3000 ng/ml 的样品分别稀释 2 倍、5 倍、10 倍,各稀释因子平行 5 个样品,LMV-12 的准确度偏差在–9.45%~7.69% 之间,精密度 RSD 在 1.49%~2.13% 之间;M4 的准确度偏差在–2.91%~7.23% 之间,精密度 RSD 在 1.53%~2.61% 之间,表明 LMV-12 或 M4 的血浆样品稀释 2~10 倍后测得结果可靠。
2.10 稳定性
LMV-12/M4 在 SD 大鼠血浆样本低、高浓度水平的样品各 3 份,室温放置 6 h,–70 ℃ 冰箱中保存 135 d,或者–70 ℃ 条件下保存 24 h 以上并经过 3 个冻融循环后,LMV-12/M4 测定结果(表 3)的准确度偏差均在标示值的 ±15% 内,符合指导原则要求。另外,研究结果显示 LMV-12/M4的血浆样品处理后的提取液于自动进样器(4 ℃)中保存 96 h 后重新测定,准确度结果均符合指导原则稳定性要求,显示样品稳定。
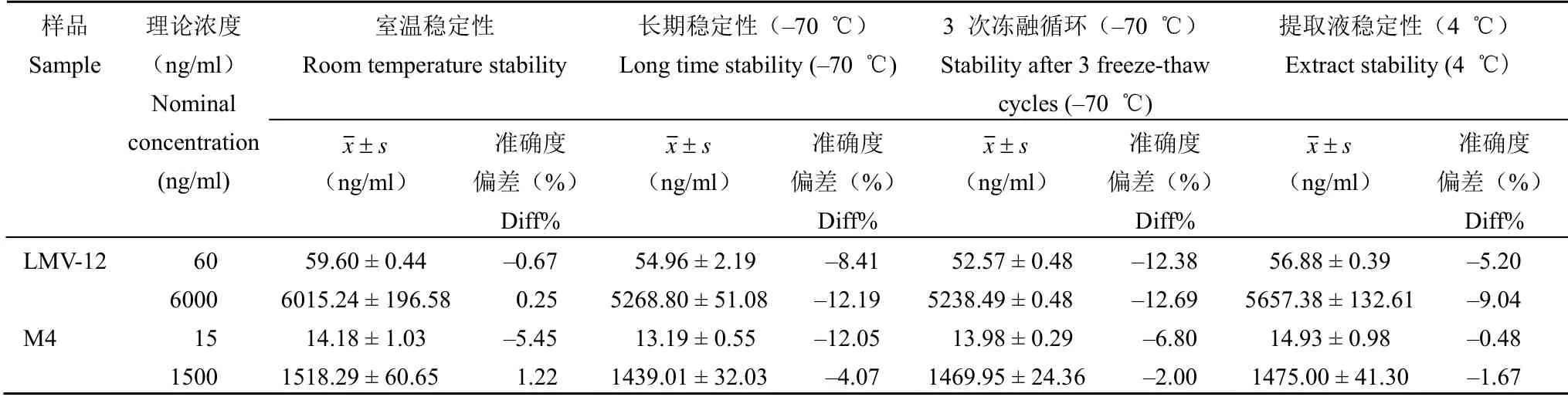
表3 SD 大鼠血浆内 LMV-12 及 M4 稳定性测试结果Table 3 Stability of LMV-12 and M4 in SD rat plasma
2.11 毒代动力学样品分析
将本方法应用于 SD 大鼠灌胃给予 LMV-12毒代动力学研究。将血浆样品按已验证方法前处理后,采用新鲜配制的标准曲线样品并新鲜建立的标准曲线,同时分析高、中、低的 QC 样品,根据当日标准曲线求算未知样品浓度和 QC 样品浓度。末次给药后 LMV-12 和 M4 的血药浓度药-时曲线见图 3,从图中可以看出,LMV-12 的Cmax为201.77 ng/ml,出现在给药后 2 h,M4 的Cmax为35.54 ng/ml,出现在给药后 4 h,比 LMV-12 的时间滞后。测试过程分析批均满足《生物样品定量分析方法验证指导原则》的要求[6-9]和毒代动力学研究的要求[1-3]。
3 讨论
本研究针对创新型抗肿瘤化合物 LMV-12,建立了采用高效液相色谱-串联三重四极杆质谱同时测定血浆中 LMV-12 原型及其代谢产物 M4 的方法,通过对样品前处理、色谱/质谱条件的优化,有效降低了残留,完整方法学验证结果显示该方法操作简单、回收率高、方法重现性好,各项验证指标均满足生物样品定量分析[6-9]的要求。进一步将本方法应用于 LMV-12 大鼠毒代动力学研究,将为本品临床前乃至临床研究提供重要信息。
猜你喜欢
系统医学(2022年6期)2022-06-13
科海故事博览·下旬刊(2022年4期)2022-05-07
口腔护理用品工业(2021年4期)2021-11-02
中国科技纵横(2019年23期)2019-02-14
科学家(2016年17期)2017-10-17
海峡科技与产业(2017年1期)2017-03-04
价值工程(2016年32期)2016-12-20
中小企业管理与科技·中旬刊(2014年10期)2015-02-03
科技视界(2015年15期)2015-01-16
