
新生儿糖尿病48例临床特征及分子遗传学分析*
2021-09-07 02:37:28梅慧芬李秀珍林云婷徐爱晶盛慧英尹曦程静张文饶敏梁翠丽刘丽
广东医学 2021年8期
梅慧芬, 李秀珍, 林云婷, 徐爱晶, 盛慧英, 尹曦, 程静, 张文, 饶敏, 梁翠丽, 刘丽
广州市妇女儿童医疗中心遗传与内分泌科(广东广州 510623)
新生儿糖尿病(neonatal diabetes mellitus,NDM)指出生后6个月内出现的持续性高血糖症,通常需要胰岛素治疗。国外报道其在活产婴儿中的发病率为1∶89 000~260 000[1-4]。根据疾病转归不同,临床上分为两种亚型:短暂性新生儿糖尿病(temporary neonatal diabetes mellitus, TNDM)和永久性新生儿糖尿病(permanent neonatal diabetes mellitus, PNDM)[5]。PNDM患者终生需要药物治疗,占NDM 大约50%~60%的TNDM在治疗几个月后会缓解,但常在青春期复发。NDM是一种遗传异质性疾病,迄今为止,已发现20多种致病基因与NDM的发病有关。不同遗传学类型NDM临床表现有很大差异,治疗方案及其预后也不同。比如大部分ATP敏感性钾离子(KATP)通道基因突变所致的NDM可以应用口服磺酰脲类药物治疗,而胰岛素(insulin,INS)基因突变者终身需要胰岛素注射等[5]。本研究总结48例NDM患儿的临床特征,并对相关致病基因进行分子遗传学研究,探讨相关遗传发病机制,为患儿制定个体化治疗方案并对家系提供遗传咨询。
1 资料与方法
1.1 一般资料 48例均为2007年2月至2019年1月于我院收治的NDM患儿。男32例,女16例,初诊年龄2~170 d。首发症状为多饮多尿32例,精神差25例,脱水并气促15例,无明显症状常规检查发现高血糖17例。2例患儿父母亲一方有糖尿病,1例患儿母亲有妊娠期糖尿病,其余无糖尿病家族史。NDM的诊断标准[6-7]:(1)生后6个月内发病;(2)符合糖尿病诊断标准;(3)病程大于2个月;(4)排除应激性高血糖、医源性或其他原因的高血糖症。符合以上4条可诊断为NDM。本研究经医院医学伦理委员会批准,并由患儿监护人签署知情同意书。
1.2 实验室检查 入院后抽血分别检测血葡萄糖、血气分析、电解质;糖化血红蛋白(采用乳胶免疫凝集抑制法,德国西门子公司试剂盒);同时做血胰岛素、C肽、游离三碘甲腺原氨酸、游离甲状腺素、促甲状腺激素、皮质醇、促肾上腺皮质激素(采用化学发光法,德国西门子公司试剂);胰岛素抗体、谷氨酸脱羧酶抗体、胰岛细胞抗体(采用免疫印迹法,深圳市亚辉龙生物科技有限公司试剂盒);胰腺影像学检查(超声或核磁共振)等。
1.3 治疗及随访 除3例血糖轻度升高的患儿,其他患儿在诊断时应用胰岛素治疗,当糖尿病酮症酸中毒缓解并且血糖稳定后,开始给予口服磺脲类降糖药格列本脲(上海信谊药厂有限公司生产,国药准字:H31020795)进行试验性替代胰岛素治疗。起始剂量每天0.2 mg/kg,分3次,根据血糖水平酌情加量,格列本脲剂量增加的幅度为每天0.1~0.2 mg/kg,胰岛素逐渐减量,如果格列本脲增加至每天1.0 mg/kg,胰岛素仍不能减量,判定为对格列本脲无反应,遂停用格列本脲,继续胰岛素治疗。由于格列本脲治疗NDM为超说明书用药,所有父母均签署了知情同意书。
临床随访时间为出院后1个月,之后每3~6个月随访1次,本研究随访至2020年12月。每次就诊时测量身高和体重,对精神运动发育进行监测,对怀疑有发育迟缓的患者进行进一步的Gesell发育量表评估。每次复诊时检测糖化血红蛋白,并进行肾功能、肝功能检查。根据检查结果及患儿的自我指尖血糖监测结果调整格列本脲或胰岛素的剂量。
1.4 分子遗传学分析 经家长知情同意后,留取患儿血液标本,采用PCR-DNA直接测序法进行KCNJ11、ABCC8、INS和葡萄糖激酶(GCK)基因全部外显子及相邻内含子测序分析,检测到变异后与人类基因突变数据库及千人基因组数据库比对,明确是否为新变异或多态性。对新错义变异进行变异位点氨基酸10种不同物种间保守性分析,并进行SIFT、PolyPhen-2软件分析预测其致病性。
对经过上面方法未检出KCNJ11、ABCC8、INS和GCK基因突变的患儿,应用甲基化特异性的多重连接依赖的探针扩增(MS-MLPA)方法检测染色体 6q24区域异常。对检出异常的患儿使用CytoScan 750k阵列(Affymetrix Inc.,Santa Clara, California)进行染色体微阵列分析(CMA)进行确认。按照规范的方法进行DNA消化、扩增、分割、标记和杂交,结果用染色体分析套件软件(Affymetrix Inc.)进行分析。
最后,对经过上述检测方法未发现分子遗传学异常的阴性样品进行二代测序全外显子检测,由诺禾致源基因检测公司完成,具体方法参照我们报道的文献[8]。
对于检测到分子遗传学异常的患儿,父母的DNA样本被进一步分析以确认其遗传性。
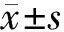
2 结果
2.1 临床特点 48例患儿(包括1对双胞胎)来自47个没有血缘关系的家庭,父母均非近亲结婚。48例中有1例胰岛细胞抗体阳性,5个月大起病时被当地误诊为1型糖尿病,5岁时因为精神运动发育迟缓、癫痫来我院就诊。除此例患儿外,所有患儿均在4个月龄大前确诊。在发病时5例患儿有贫血;5例有精神运动发育迟缓;2例有先天性喉喘鸣,2例有皮肤血管瘤;2例有特殊面容;2例有巨舌;分别伴有房间隔缺损、室间隔缺损或脐疝各1例。
48例患儿中有30例(62.5%)为PNDM,18(37.5%)为TNDM。PNDM和TNDM的临床特点见表1~2,两种类型NDM在男女比例、孕龄、出生体重和起病时的血糖方面差异无统计学意义(P>0.05)。然而,与PNDM相比,TNDM起病的时间更早(P=0.015),TNDM发病时糖尿病酮症酸中毒(diabetes ketoacidosis,DKA)发生率较低(P=0.03)。TNDM缓解年龄2.5~10个月,随访时间2.7~12.2年,其中4例复发,复发年龄4.5~8岁。
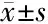
表1 PNDM 与TNDM临床特点比较
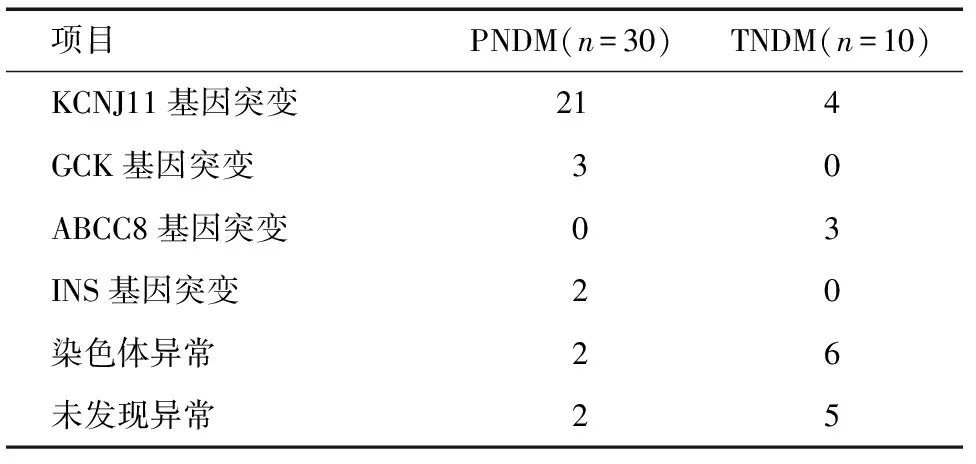
表2 分子遗传学结果 例
2.2 分子遗传学检测结果 48例中有41例(85.4%)明确了NDM的分子遗传学病因(表3~5),其中有25例(50.0%)、3例(6.3%)、2例(4.2%)、3例(6.3%)、8例(16.7%)分别检出KCNJ11、ABCC8、INS、GCK及染色体异常(其中染色体 6q24区域异常6例)。1例ABCC8复合杂合突变(p.Gly251Glu,p.Ile467Phe)及1例KCNJ11杂合突变(p.Ala18Gly)为新发突变。
表3 48例NDM患儿的临床特点
在30例PNDM病例中,KCNJ11基因突变是最常见的原因,占70.0%,其中5例 p.Arg201His(R201H)突变,5例p.Val59Met(V59M)突变,4例p.Arg201Cys(R201C)突变,2例p.Cys42Arg(C42R)突变,其他类型突变各1例,另外有3例检出GCK基因突变,2例检出INS基因突变,1例为6q24染色体异常,1例为染色体17p13.3微重复。尚有2例未检出分子遗传学异常。
在18例TNDM中,4例KCNJ11基因突变所致,3例ABCC8基因突变所致,5例染色体6q24异常,1例检出染色体1p36.23p36.12微缺失。尚有5例未检出分子遗传学异常。
2.3 治疗及随诊 48例患儿中有41例尝试从胰岛素过渡到口服格列本脲治疗,其中28例(28/41,69%)成功转用格列本脲单药治疗,且血糖更加平稳,低血糖发生率也显著减少。停用胰岛素时格列本脲的剂量为0.3~0.8 mg/(kg·d),平均(0.54±0.19)mg/(kg·d)。41例患儿试用格列苯脲过程中有6例发生短暂喂养不良,2例出现短暂性腹泻。28例格列本脲治疗有效的NDM患儿中,有8例在3~10个月时血糖正常,其余20例患儿需要长期服用格列本脲至今,随访1.8~8.6年,平均(5.6±4.3)年,未发现不良反应(如牙齿发黄、胃肠道不良反应、肾功能和肝功能障碍等)。除1例KCNJ11突变(p.V64M)患儿血糖及糖化血糖偏高,在1岁左右需要联合应用小剂量胰岛素外,其他19例患儿只用格列本脲治疗,血糖水平及糖化血红蛋白水平均接近正常,目前格列本脲的维持剂量为0.2~0.4 mg/(kg·d),平均(0.30±0.07) mg/(kg·d),末次糖化血红蛋白为5.2%~6.2%,平均(5.82±0.25)%。这28例格列本脲治疗治疗有效的NDM患儿的基因突变类型详见表4。
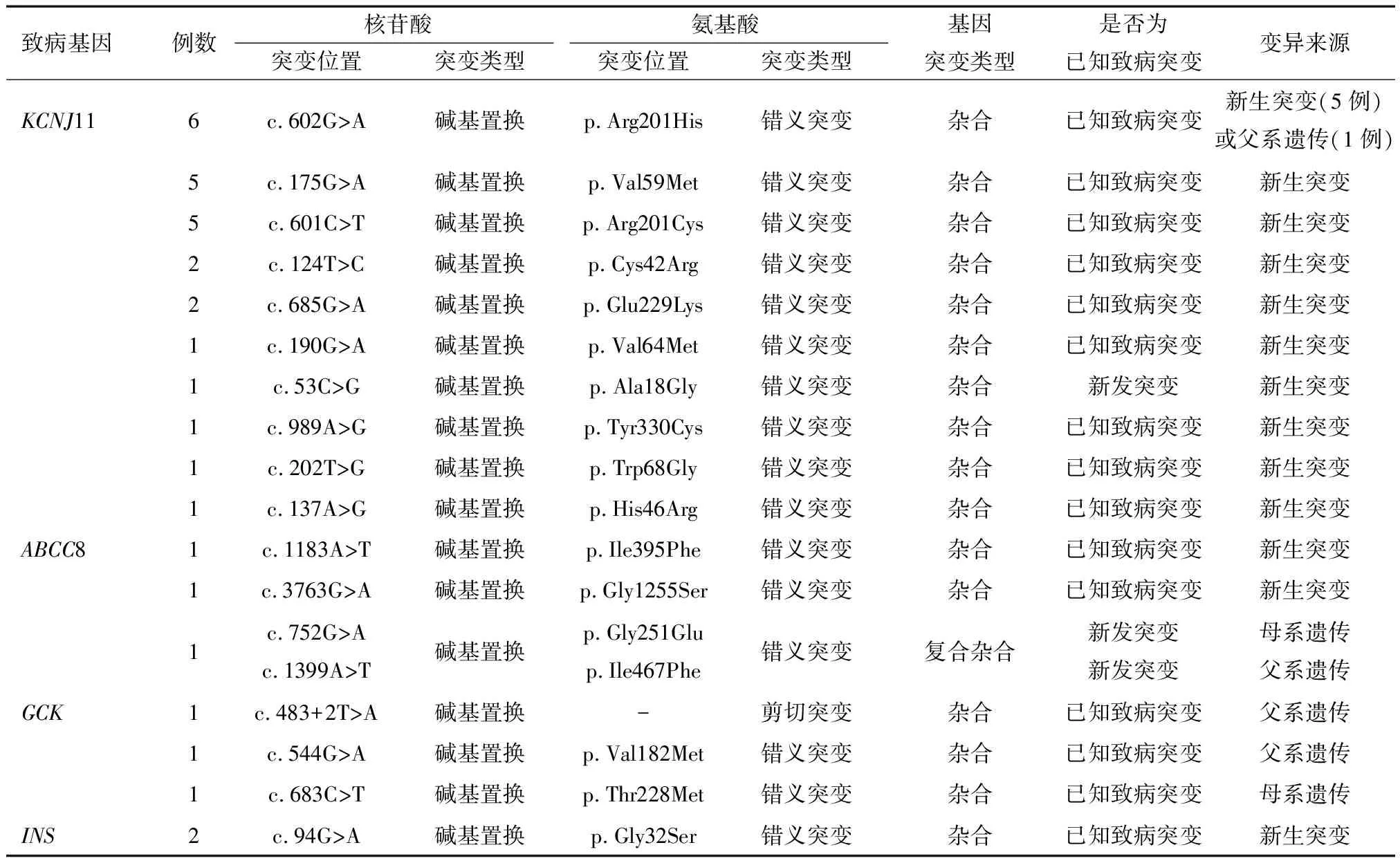
表4 41例NDM患儿分子遗传学结果
48例患儿随访至2020年12月,6例KCNJ11基因突变患儿有轻度精神运动发育迟缓(5例V59M突变,1例V64M突变),1例染色体 1p36.23p36.12微缺失患儿有中度精神运动发育迟缓,1例GCK基因突变患儿诊断为家族性矮小症。
3 讨论
目前NDM的分型是基于临床随访期间的病情是否缓解分为PNDM和TNDM,随诊至18个月大时,血糖正常、糖化血红蛋白正常且无需胰岛素或口服降糖药物治疗可诊断为TNDM;如果仍然需要胰岛素或口服降糖药来维持正常血糖者为PNDM[9-10]。TNDM与PNDM在发病初期难以区分。国外文献报道研究超过一半以上的NDM为PNDM[11]。本研究48例患儿中,62.5%(30/48)是PNDM,PNDM与TNDM在出生体重、起病时血糖平均水平无明显差异,但TNDM发病年龄更早(P=0.015),DKA发病率更低(P=0.03)。TNDM缓解年龄2.5~8个月,其中4例出现复发,复发年龄4.5~8岁。国外文献报道大多数TNDM在儿童晚期或者青春期会持续糖耐量受损或者是糖尿病复发。糖尿病复发原因尚不清楚,可能与青春期胰岛素抵抗有关的。本研究中4例患儿复发年龄较文献报道相对偏早,其原因有待进一步研究。
NDM是单基因遗传变异导致的特殊类型糖尿病,明确分子遗传学诊断使精准治疗成为可能,并且有助于评估预后及对家族进行遗传咨询。PNDM的病因较为多样化,近年来国外研究显示半数以上的PNDM由编码胰岛β细胞KATP通道的内向整流6.2 (inwardly rectifying potassium channels,Kir6.2)亚单位和磺脲类受体1(sulfonylurea receptor 1,SUR1)亚单位的KCNJ11和ABCC8基因突变所致,其他致病基因有INS基因突变、GCK基因突变等。染色体6q24区域异常是TNDM的主要病因,目前已发现的6号染色体异常包括以下3种情况[12]:(1)6号染色体的父源性单亲二倍体(paternal uniparental disomy of chromosome 6,UPD6),已经证实与散发的TNDM发病相关。(2)父源性6号染色体长臂的部分复制,当家系中出现这种不平衡复制时,仅有来自父亲的异常复制才会发病,提示这一缺陷可能存在于印迹区。(3)少数患儿是由于染色体6q24关键基因片段的甲基化缺失。KATP通道基因突变是导致TNDM的第二大原因,其余类型TNDM临床少见。

表5 染色体异常检测结果
本研究48例NDM中有41例(85.4%)检测到分子遗传学异常,其中KCNJ11(60.9%)和ABCC8(7.3%)突变最为常见。6q24染色体异常(12.2%)是另一常见的病因。我们的发现与日本和欧洲国家的研究结果相似[1,6,13]。在30例PNDM病例中,KCNJ11基因突变是最常见的原因(占70.0%)。其中5例 R201H突变, 5例V59M突变, 4例 R201C突变,2例C42R突变,其他类型突变各1例,均为已经报道的致病突变。国外研究显示KCNJ11基因变异引起的NDM表型与基因突变位点相关,位于Kir6.2亚基ATP结合区域的变异多表现为单纯的NDM;而远离ATP结合区域位点的变异出现DEND综合征(包括发育迟缓、癫痫、NDM)或部分性DEND综合征,有发育迟缓和NDM,但无癫痫。目前报道有出现神经系统症状的基因型包括V59M、R201C、G53D、R50P、R50G、C166Y、I167L、G344等,其中以V59M突变最为常见[13]。本研究5例V59M突变NDM中,4例为部分性DEND综合征,1例为DEND综合征;另外1例V64M突变患儿为部分性DEND综合征;其他类型突变患儿未出现神经系统异常临床表型。最近有研究[14]发现少数KCNJ11基因变异引起的NDM患儿会出现发育性共济障碍如视觉空间障碍、注意力缺陷多动障碍等,本组患儿中未观察到类似的神经系统表现,估计与年龄尚小有关,有待进一步观察。6q24染色体异常通常导致TNDM,但本研究中1例6q24染色体异常患儿为PNDM,提示对PNDM患儿也有必要进行染色体6q24异常相关检查。与PNDM发病相关的其他基因包括GCK基因及INS基因等,INS基因突变所致的患儿起病年龄往往较晚,大部分患儿6个月后才发病。
在18例TNDM中,KCNJ11(4/18)和ABCC8(3/18)基因突变是主要原因,约占TNDM的38.9%。6q24染色体的遗传异常占27.8%(5/18),而在欧美人群中,6q24染色体异常是TNDM最常见的原因[15]。鉴于KCNJ11在NDM中的高突变率和高度相关基因型和表型,建议在NDM患儿中首先筛查KCNJ11突变,然后筛查ABCC8或其他与特定临床表现相关的基因。当然,有条件者可以直接行二代测序检查。
KATP通道是调节胰岛素分泌的关键因素,在生理情况下,血糖升高后,葡萄糖被转运至胰岛β细胞,在GCK等关键酶作用下经过三羧酸循环,细胞内ATP浓度升高,刺激Kir6.2通道关闭,细胞膜去极化,胞膜上电压依赖性Ca2+通道开放,Ca2+内流,从而引起胰岛素分泌。当编码胰岛β细胞KATP通道的亚单位Kir6.2或SURl亚单位的KCNJ11基因和ABCC8基因发生杂合子激活突变时,KATP通道与细胞内ATP亲和力下降,在葡萄糖刺激下无法正常关闭,从而导致B细胞内胰岛素无法正常释放。文献报道90%以上KCNJ11基因突变及85%以上ABCC8基因突变所致的NDM均可从胰岛素治疗转变到磺酰脲类药物治疗。磺脲类降糖药物可以直接作用于SUR1,使KATP通道关闭,从而使胰岛素得以正常释放;同时,由于SUR广泛分布于神经细胞、骨骼肌细胞等胰腺外组织,磺脲类药物对基因缺陷所导致的其他伴随症状如精神运动发育迟缓、癫痫等也有明显的改善作用。另外研究报道磺脲类降糖药物对部分6q24染色体异常NDM也有效果[16]。
本研究41例NDM中,有69%患儿能成功转用格列本脲,且葡萄糖更稳定,低血糖发生率也更少。有趣的是,本研究中有1例1p36缺失综合征患儿在49 d时应用格列本脲治疗反应良好,其机制有待进一步研究。
本研究中格列本脲的维持剂量<0.5 mg/(kg·d)。长期随访没有出现严重的不良反应,一些可能与药物相关的不良反应是轻度喂养困难或短暂性腹泻,所有患者均未因不良反应而停止治疗,体格发育与同龄儿相仿。我们的研究结果显示磺酰脲类药物治疗NDM是安全有效的,与国外研究一致,提示磺酰脲类药物是治疗NDM的一种有效方法。此外,早期磺酰脲类药物治疗可以改善神经发育障碍。本研究5例伴有精神运动发育迟缓患儿早期应用格列本脲治疗,不但血糖控制更理想,同时神经系统症状均有改善。因此,磺脲类降糖药物比外源性胰岛素注射更加符合这类NDM患者的生理需要。
磺酰脲类药物转化成功与否与KATP通道突变类型的有关。目前已发现KCNJ11 基因70多种不同的突变,均为杂合突变,其中R201H、V59M 为热点突变,可用磺脲类药物替换胰岛素治疗的患儿的突变位点主要为R201H、V59M、R201C、F35V、H46Y、R50Q、G53N、G53R和K170T等,而仍然需要依赖胰岛素治疗者突变位点为Q52R、L164P 和I296L等[17]。本研究中24例KCNJ11突变的患儿试用格列本脲替换胰岛素治疗,23例(95.8%)成功停用了胰岛素,仅1例C42R突变患儿格列苯脲治疗(诊断年龄115 d,合并DKA,转换时年龄125 d)无效,但另1例携带同样突变的患儿(诊断年龄4 d,无合并DKA,转换时年龄18 d)仅需要小剂量格列苯脲治疗,血糖就接近正常,提示磺酰脲类药物转化成功与否还受其他因素的影响。国外研究显示年龄越小、病程越短的患儿转用磺酰脲类药物的成功率越高[18]。较早开始使用磺酰脲类药物治疗的患儿对磺酰脲类药物的更敏感,也可能导致较低的维持剂量[19-20]。随着年龄的增长或糖尿病病程的延长,对磺酰脲类药物的敏感性下降,可能是由于使用胰岛素治疗的患者随着时间的推移,胰岛β细胞数量逐渐减少。本研究41例患儿尝试了应用格列本脲替换胰岛素治疗,其中28例(69%)成功停用了胰岛素,血糖水平更加稳定。目前格列本脲的维持治疗平均剂量为(0.3±0.07) mg/(kg·d),与Bowman等[21]的研究相接近。与注射胰岛素相比,口服格列本脲可以降低医疗成本,使血糖水平更稳定。此外,口服药物可以避免注射的痛苦,提高家庭生活质量。
综上所述,本研究显示大部分(85.4%)NDM患儿检测到分子遗传学异常,其中KCNJ11和ABCC8突变最为常见,6q24染色体异常是另一常见的病因;早期使用格列本脲治疗是安全有效的。
猜你喜欢
药物与人(2023年1期)2024-01-15 03:32:15
现代农药(2022年2期)2022-04-11 11:53:42
山东医药(2020年25期)2020-12-29 11:36:52
福建基础教育研究(2019年10期)2019-05-28 08:27:04
求学·理科版(2017年3期)2017-04-27 22:06:36
中国药业(2017年6期)2017-04-26 04:02:31
法制博览(2016年11期)2016-11-14 10:28:42
药学研究(2015年7期)2015-03-14 11:48:20
刑事技术(2015年5期)2015-02-26 23:57:51
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:43
