
新型非共价键构象锁定型小分子的合成及性能研究
2021-09-01 12:45李宇翔王英英秦红梅杨建业
合成化学 2021年8期
王 梅, 李宇翔, 王英英, 秦红梅, 杨建业
(西安科技大学 材料科学与工程学院,陕西 西安 710054)
近年来,有机稠环化合物因具有刚性的共平面结构和强π-π相互作用等优势,在有机半导体领域展现出良好的应用前景[1-2]。其中,稠环化合物研究在有机光伏领域取得了突破性进展,光电转换效率超过了18%[3-4]。然而,稠环类材料合成难度较大、价格昂贵等缺点限制了其进一步商业应用[5]。为解决这一难题,研究人员提出用“非共价键构象锁”的方法构建类稠环有机化合物,即通过在分子内引入S…O和O…H等非共价键作用力来阻止单键旋转,从而提高有机分子的共平面性,扩大分子的共轭长度[6-7]。基于这种方法,科研工作者已经开发出多种高效有机半导体材料,如IEICO[8], IDT-Tz[9], IDT-BOC6[10], IDTOT2F[11],PhIC[12], PTICH[13]等。
鉴于此,本文采用“非共价键构象锁”策略,通过Stille偶联,Knoevenagel缩合等反应合成了两个新型小分子化合物PTIC和PTTIC(Scheme 1)。采用UV-vis、 CV、 DSC及掠入射广角X射线散射(GIWAXS)对其结构和性质进行了表征。

Scheme 1
1 实验部分
1.1 仪器与试剂
BRUKER 400 MHz型核磁共振仪(TMS为内标);Shimadzu UV-3600 Plus型紫外-可见光谱仪;CHI 600E型电化学工作站;Q200 V24.4型差示扫描量热仪;PLS-II 9A U-SAXS 型掠入射广角X-射线散射仪。
化合物M1,T1,M2和T2参考文献方法[14-15]合成;双氰基茚满二酮,朔纶有机光电科技(北京)有限公司;甲苯使用前经金属钠处理;其余所用试剂均为分析纯。
1.2 合成
(1) 化合物M3和T3的合成(以M3为例)
将化合物M1(1.00 g, 1.39 mmol)、M2(1.45 g, 3.91 mmol)和催化剂Pd(PPh3)4(0.23 g, 0.02 mmol)加入15 mL甲苯中,氮气保护下,于120 ℃反应36 h。用二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,滤液减压除溶,粗产物经硅胶柱层析(洗脱剂:石油醚)纯化得油状液体M30.95 g,收率95%;1H NMR(400 MHz, CDCl3)δ: 7.52(d,J=3.6 Hz, 1H), 7.32(d,J=5.1 Hz, 1H), 7.24(s, 1H), 7.11~7.06(m, 1H), 3.96(d,J=5.5 Hz, 2H), 1.88(t,J=6.1 Hz, 2H), 1.53~1.51(m, 1H), 1.33~1.08(m, 24H), 0.86(s, 6H)。
用类似的方法合成T3,收率76%;1H NMR(400 MHz, CDCl3)δ: 7.72(s, 1H), 7.26(s, 1H), 6.96(s, 1H), 4.00(d,J=5.6 Hz, 2H), 2.75~2.70(m, 2H), 1.92~1.88(m, 1H), 1.81~1.77(m, 2H), 1.36~1.24(m, 46H), 0.94~0.85(m, 9H)。
(2) 化合物M4和T4的合成
氮气保护下,将化合物M3(1.20 g, 1.66 mmol)溶解于四氢呋喃(40 mL)溶液中,冷却至-78 ℃,缓慢滴加正丁基锂(1.73 mL, 4.14 mmol),滴毕,反应2 h。恢复至室温,滴加DMF(0.36 g, 4.97 mmol),反应12 h。用水淬灭反应,二氯甲烷萃取,有机相用无水硫酸镁干燥,过滤,滤液减压除溶,粗产品经过硅胶柱层析(洗脱剂:二氯甲烷/石油醚=1/9,V/V)纯化得M40.83 g,收率52%;1H NMR(400 MHz, CDCl3)δ: 9.93(s, 1H), 7.75(d,J=4.0 Hz, 1H), 7.66(d,J=4.0 Hz, 1H), 7.31(s, 1H), 4.01(d,J=5.6 Hz, 2H), 1.93(s, 1H), 1.34~ 1.26(m, 24H), 0.88~0.85(m, 6H)。
氮气保护下,将无水DMF(0.70 mL)加入双口瓶中,在0 ℃下加入三氯氧磷(1.43 mL, 17.4 mmol),搅拌反应1 h。将化合物T3(1.20 g, 1.02 mmol)溶解于1,2-二氯乙烷(DCE, 15 mL)溶液中,滴加到DMF和三氯氧磷体系中,滴毕,搅拌20 min;加热至90 ℃,反应12 h。用二氯甲烷萃取,合并有机相,无水硫酸镁干燥,减压除溶,粗产品经硅胶柱层析(洗脱剂:二氯甲烷/石油醚=2/8,V/V)纯化得T4,收率68%;1H NMR(400 MHz, CDCl3)δ: 10.10(s, 1H), 7.75(s, 1H), 7.27(s, 1H), 4.02(s, 2H), 2.12(s, 2H), 1.92(s, 1H), 1.86(s, 2H), 1.45~1.16(m, 46H), 0.87( d,J=6.2 Hz, 9H)。
(3) 化合物PTIC和PTTIC的合成(以PTIC为例)
氮气保护下,将化合物M4(0.20 g, 0.26 mmol)溶于氯仿(15 mL)中,加入双氰基茚满二酮(IC)(0.19 g, 1.02 mmol)和吡啶(0.40 mL),于65 ℃回流反应12 h。冷却至室温,倒入甲醇中,过滤,滤饼依次用甲醇和丙酮洗涤,经硅胶柱层析(洗脱剂:二氯甲烷/石油醚=3/7,V/V)纯化得深蓝色固体PTIC0.17 g,收率58.6%;1H NMR(400 MHz, CDCl3)δ: 8.88(s, 1H), 8.71(dd,J=6.5 Hz, 1.6 Hz, 1H), 7.93(dt,J=8.1 Hz, 5.3 Hz, 2H), 7.82~7.72(m, 3H), 7.40(s, 1H), 4.12(d,J=5.2 Hz, 2H), 2.06(dt,J=11.9, 5.8 Hz, 1H), 1.71~1.11(m, 24H), 0.83(t,J=6.8 Hz, 6H);13C NMR(100 MHz, CDCl3)δ:187.84, 160.84, 155.70, 150.70, 144.53, 139.91, 137.83, 137.76, 136.97, 135.04, 134.41, 127.83, 125.28, 124.15, 123.70, 122.95, 114.55, 114.43, 112.39, 72.82, 69.62, 59.48, 38.14, 31.88, 31.85, 31.43, 31.19, 30.01, 29.67, 29.61, 29.33, 26.90, 22.65, 22.63, 14.07。
用类似的方法合成深蓝色固体PTTIC,收率62.3%;1H NMR(400 MHz, CDCl3)δ: 9.03(s, 2H), 8.62(s, 2H), 7.95~7.92(m, 2H), 7.75(t, 6H), 7.25(s, 2H), 4.10(d,J=4.9 Hz, 4H), 3.14(t,J=7.6 Hz, 4H), 1.99(d,J=7.9 Hz, 4H), 1.81(q,J=8.6 Hz, 7.7 Hz, 4H), 1.48~1.41(m, 8H), 1.27(m, 76H), 0.89~0.82(m, 18H);13C NMR(100 MHz, CDCl3)δ:187.71, 160.72, 155.15, 150.77, 144.60, 144.11, 139.98, 137.91, 137.80, 137.04, 135.10, 134.48, 127.91, 127.04, 125.34, 124.23, 123.77, 123.03, 121.53, 114.61, 114.48, 112.50, 72.87, 69.69, 38.20, 31.93, 31.90, 31.49, 30.06, 29.65, 29.38, 26.98, 26.95, 22.70, 22.68, 14.11。
2 结果与讨论
2.1 光学性能
PTIC和PTTIC在三氯甲烷溶液和薄膜状态下的紫外吸收谱图见图1。从图1可以看出,在溶液状态下,两种小分子在500~700 nm内均具有较宽和较强的光谱吸收,PTIC和PTTIC的最大吸收峰分别位于621 nm和647 nm,其摩尔吸光系数分别为7.64×104M-1cm-1和1.40×105M-1cm-1,表明并噻吩为π桥的PTTIC具有更好的光吸收性能。在固体薄膜状态下,PTIC和PTTIC最大吸收峰均出现明显的红移(~77 nm),且在640~670 nm出现明显肩峰,表明在薄膜状态下,分子间可形成更有序的J聚集[16]。相对于单个噻吩作为π桥的PTIC,并噻吩为π桥的PTTIC在溶液态和薄膜态下最大吸收红移程度均超过24 nm,表明PTTIC具有更好的共轭性和更强的π-π堆积。由薄膜状态的起始吸收波长(λonset)计算得到两种小分子光学带隙Egopt分别为1.61 eV和1.56 eV,具体数据见表1。

表1 小分子材料的光学和电化学性能参数

λ/nm
2.2 电化学性能
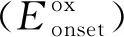

Potential(V Vs Ag/Ag+)
2.3 热力学性能
因PTIC和PTTIC结构相似,仅对PTIC采用差示扫描量热法(DSC)进行了热力学性能研究。图3为PTIC的DSC曲线。从图3可以看出,PTIC在加热状态下于262 ℃出现一个尖锐吸热峰,在冷却状态下于236 ℃也出现明显放热峰,表明PTIC具有很好的结晶性,这有利于分子的电荷分离和传输[18]。
Temperature/℃
2.4 GIWAXS表征结果
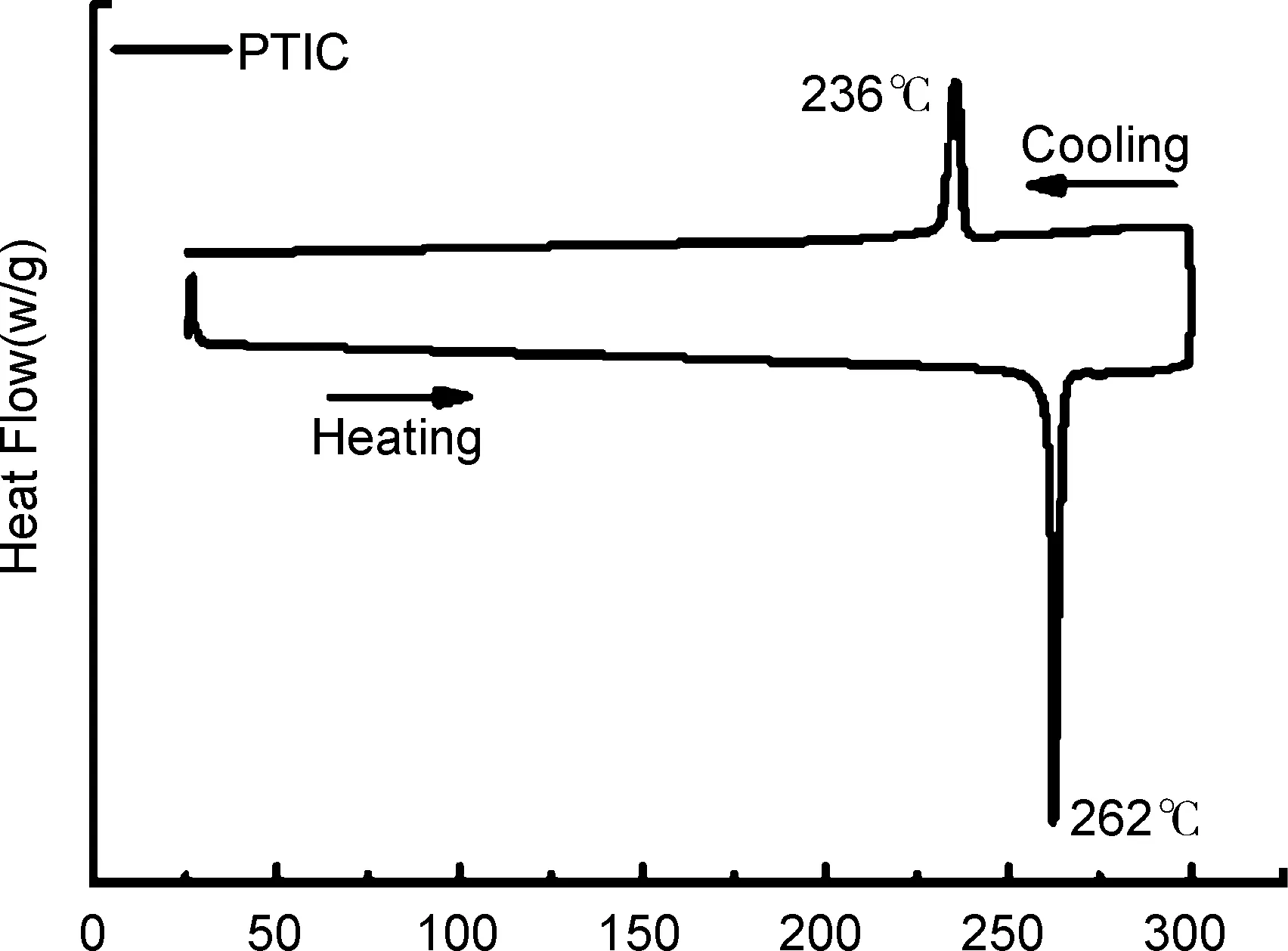
为了进一步研究小分子的结晶性和取向性,采用GIWAXS对PTIC进行表征,结果见图4。由图4(a)和(b)可知,PTIC纯膜在面外(OOP)方向上qz=0.36、 0.73、 1.08和1.44 Å-1处有明显的(100)、 (200)、 (300)和(400)的层状方向(Lamellar)的衍射峰,堆积距离分别为17.5、 8.6、 5.8和4.4 Å,由此可以判断PTIC纯膜更趋向于Edge on的堆积取向,这有利于层状方向上的电荷转移。用Scherer方程[19]计算得到(100)Lamellar堆积峰在OOP方向的结晶相干长度(CCL100)为169.6 Å,说明PTIC在制备成薄膜后表现出较强的结晶性,在层状方向上排列高度有序,有助于改善分子的电子传输能力。
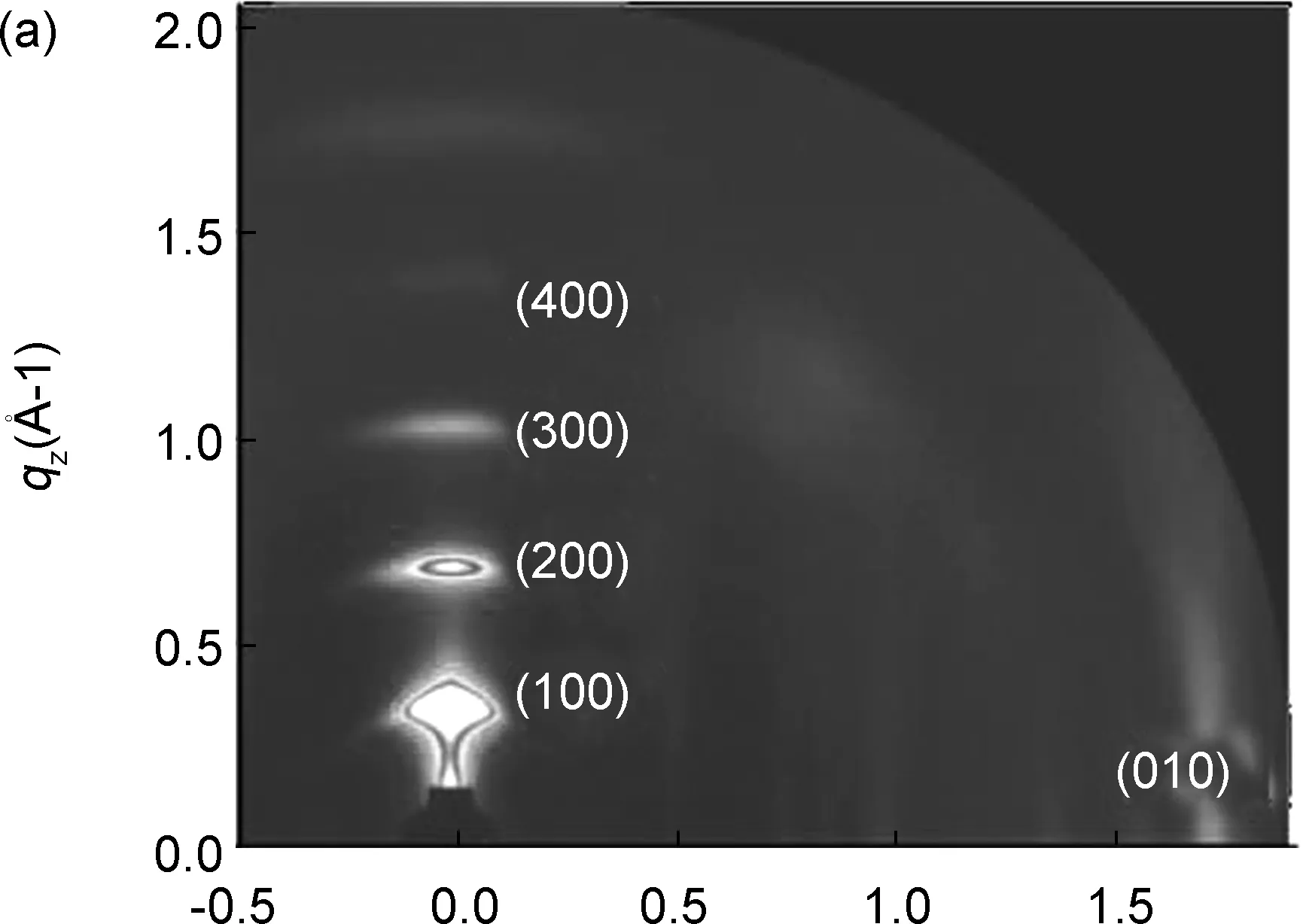
qxy/Å-1
以烷氧基苯环为核心,分别以单噻吩和并噻吩作为π桥,选择吸电子基团双氰基茚满二酮(IC)为端基,通过Stille偶联和诺文格尔缩合等反应合成了两个新型小分子PTIC和PTTIC。性能测试结果表明,PTIC和PTTIC具有良好的光学性能和结晶性能。分子内S…O和O…H作用形成非共价键构象锁增加了分子有效共轭长度,整体表现出良好的主链平面性和刚性,有利于分子内电荷转移。
猜你喜欢
武汉工程大学学报(2022年4期)2022-08-26
化学工程师(2022年3期)2022-04-19
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
鞍钢技术(2020年5期)2020-10-10
吉林农业(2019年6期)2019-06-11
中国医药导报(2019年1期)2019-03-14
教育教学论坛(2018年38期)2018-09-25
分析化学(2017年12期)2017-12-25
江苏农业科学(2016年6期)2016-07-25
科技与创新(2015年20期)2015-10-29
