
视网膜常染色体显性遗传病的特点及基因治疗策略
2021-08-23 02:30刘彦博综述庞继景审校
中华实验眼科杂志 2021年8期
刘彦博 综述 庞继景 审校
1厦门大学医学院 厦门大学眼科研究所 福建省眼科与视光科学重点实验室 361000;2厦门大学附属厦门眼科中心 361000
遗传性视网膜疾病(inherited retinal diseases,IRDs)是临床上常见且危害严重的眼科遗传疾病,由于光感受器细胞营养不良、功能障碍或过早凋亡而导致视力丧失[1]。IRDs为主要致盲眼病之一,具有高度遗传异质性,全世界患病率约为1/2 000[2],常见的IRDs包括视网膜色素变性(retinitis pigmentosa,RP)、无脉络膜症(choroidoremia,CHM)、先天性黑矇(Leber congenital amaurosis,LCA)、先天性静止性夜盲(congenital stationary night blindness,CSNB)、视锥或视锥-视杆细胞营养不良(cone or cone-rod dystrophy,CD/CRD)、青少年黄斑变性(Stargardt disease,STGD)等,目前已发现270多个致病基因[3],国内尚缺乏相关治疗药物。
1 IRDs主要类型
IRDs根据遗传方式可分为常染色体显性遗传、常染色体隐性遗传、性染色体连锁遗传、线粒体遗传以及双基因遗传5种主要类型[4]。(1)常染色体遗传 常染色体遗传是由染色体结构或数目异常引起的一类疾病并且致病基因突变出现在常染色体上,分为常染色体显性遗传和常染色体隐性遗传。(2)性染色体连锁遗传 性染色体连锁遗传是指由位于性染色体上的致病基因控制的性状随性别而遗传的现象,这些疾病在男女中的发病概率存在明显差异。人类的性连锁遗传包括X连锁遗传和Y连锁遗传,X连锁遗传病较Y连锁遗传病多见。X连锁遗传根据基因显隐性的不同,分为X连锁显性遗传病和X连锁隐性遗传病。X连锁显性遗传的病种较少,但只要存在致病基因就会发病,通常男性患者的病情较重,女性多为杂合子,发病率却是男性的2倍。相反X连锁隐性遗传病的男性患者更多,其与正常女性婚育一般不会有患病子女,但女儿都是致病基因携带者[5]。(3)线粒体遗传 线粒体是真核细胞中一类重要的细胞器,是细胞氧化磷酸化产生ATP的主要场所,线粒体DNA(mitochondrial DNA,mtDNA)是线粒体中的遗传物质,这些基因的突变能引起线粒体疾病。线粒体基因变异的表型依赖于细胞质内突变型和野生型mtDNA的相对比例,只有当突变达到一定阈值时,才会引起特定组织器官功能的障碍,表现出临床症状[6]。由于线粒体主要通过卵细胞传递,因此线粒体基因组的遗传方式不符合孟德尔遗传规律,而是遵循母系遗传的规律[7-9]。先证者的父亲一般不会有携带致病突变的风险;其母亲通常携带突变型mtDNA,但由于其细胞质内突变的mtDNA可能未达到阈值或在某种程度上受核影响而不一定发病;先证者同胞的风险取决于其母亲的状况,如果母亲携带突变的mtDNA,先证者的所有同胞将遗传该突变,但有些可能没有临床症状;先证者后代是否携带mtDNA取决于先证者性别,携带突变mtDNA的男性一般不会将突变遗传给下一代,而携带突变mtDNA的女性不管发病与否,都会将突变遗传给后代。Leber遗传性视神经病是常见的由线粒体功能障碍引起的视神经病变,属于视神经退变性母系遗传病[10]。(4)双基因遗传 对于双基因遗传目前没有明确的定义,最常见的定义为通过2个基因位点组成的基因型能够比1个基因位点组成的基因型更清楚地解释部分患者和他们未患病(影响程度更轻)的亲属表型[11],如尤塞综合征中2个不同基因同时突变时才产生临床表型。在OMIM(Online Mendelian Inheritance In Man)中,双基因遗传造成临床表型主要分为以下2种情况:1)双基因显性遗传,是指由2个不同基因的杂合子突变;2)双基因隐性遗传,是指第1个基因发生了纯合或复合杂合突变,第2个基因只发生了1个等位基因的杂合突变[12]。
2 常染色体显性IRDs及其相关基因
常染色体显性遗传是指控制性状或疾病的显性基因位于常染色体的遗传方式,这种遗传方式控制的疾病称为常染色体显性遗传病[13]。当常染色体2个等位基因只有1个正常时,称为杂合子。在杂合子状态下,如果表达出相应的临床症状和体征,则为显性遗传;如果需要2个等位基因都发生突变时才会发病,则称为隐性遗传。典型的常染色体显性遗传病系谱往往具备共同的特点:(1)患者双亲之一患病,且多为杂合体;(2)患者同胞有1/2可能患病;(3)患者后代有1/2可能患病;(4)男女患病机会均等;(5)连续传递[14]。常染色体显性IRDs包括脉络膜视网膜萎缩(chorioretinal atrophy,CA)、CD/CRD、CSNB、LCA、黄斑变性(macular degeneration,MD)、RP、视网膜发育疾病(ocular-retinal developmental disease,ORD)、视神经萎缩(optic atrophy,OA)、伴有视网膜变性的综合性疾病及其他视网膜病变,其相关致病基因见表1(数据来源:https://sph.uth.edu/retnet/),不同形式常染色体显性IRDs之间存在相同的致病基因(图1)。

表1 常染色体显性IRDs相关基因常染色体显性IRDs分类相关致病基因CAPRDM13、RGR、TEAD1CD/CRDAIPL1、CRX、GUCA1A、GUCY2D、PITPNM3、PROM1、PRPH2、RIMS1、SEMA4A、UNC119CSNBGNAT1、PDE6B、RHOLCACRX、IMPDH1、OTX2MDBEST1、C1OTNF5、CTNNA1、EFEMP1、ELOVL4、FSCN2、GUCA1B、HMCN1、IMPG1、OTX2、PRDM13、PROM1、PRPH2、RP1L1、TIMP3RPADIPOR1、ARL3、BEST1、CA4、CRX、FSCN2、GUCA1B、HK1、IMPDH1、IMPG1、KLHL7、NR2E3、NRL、PRPF3、PRPF4、PRPF6、PRPF8、PRPF31、PRPH2、RDH12、RHO、ROM1、RP1、RP9、RPE65、SAG、SEMA4A、SNRNP200、SPP2、TOPORSORDVCANOAAFG3L2、MFN2、NR2F1、OPA1伴有视网膜变性的综合性疾病ABCC6、AFG3L2、ATXN7、COL11A1、COL2A1、JAG1、KCNj13、KIF11、MFN2、OPA3、PAX2、TREX1、VCAN其他视网膜病变BEST1、CAPN5、CRB1、ELOVL1、FZD4、ITM2B、LRP5、MAPKAPK3、MIR204、OPN1SW、RB1、RCBTB1、TS-PAN12、ZNF408 注:IRDs:遗传性视网膜疾病;CA:脉络膜视网膜萎缩;CD/CRD:视锥或视锥-视杆细胞营养不良;CSNB:先天性静止性夜盲;LCA:先天性黑矇;MD:黄斑变性;RP:视网膜色素变性;ORD:视网膜发育疾病;OA:视神经萎缩
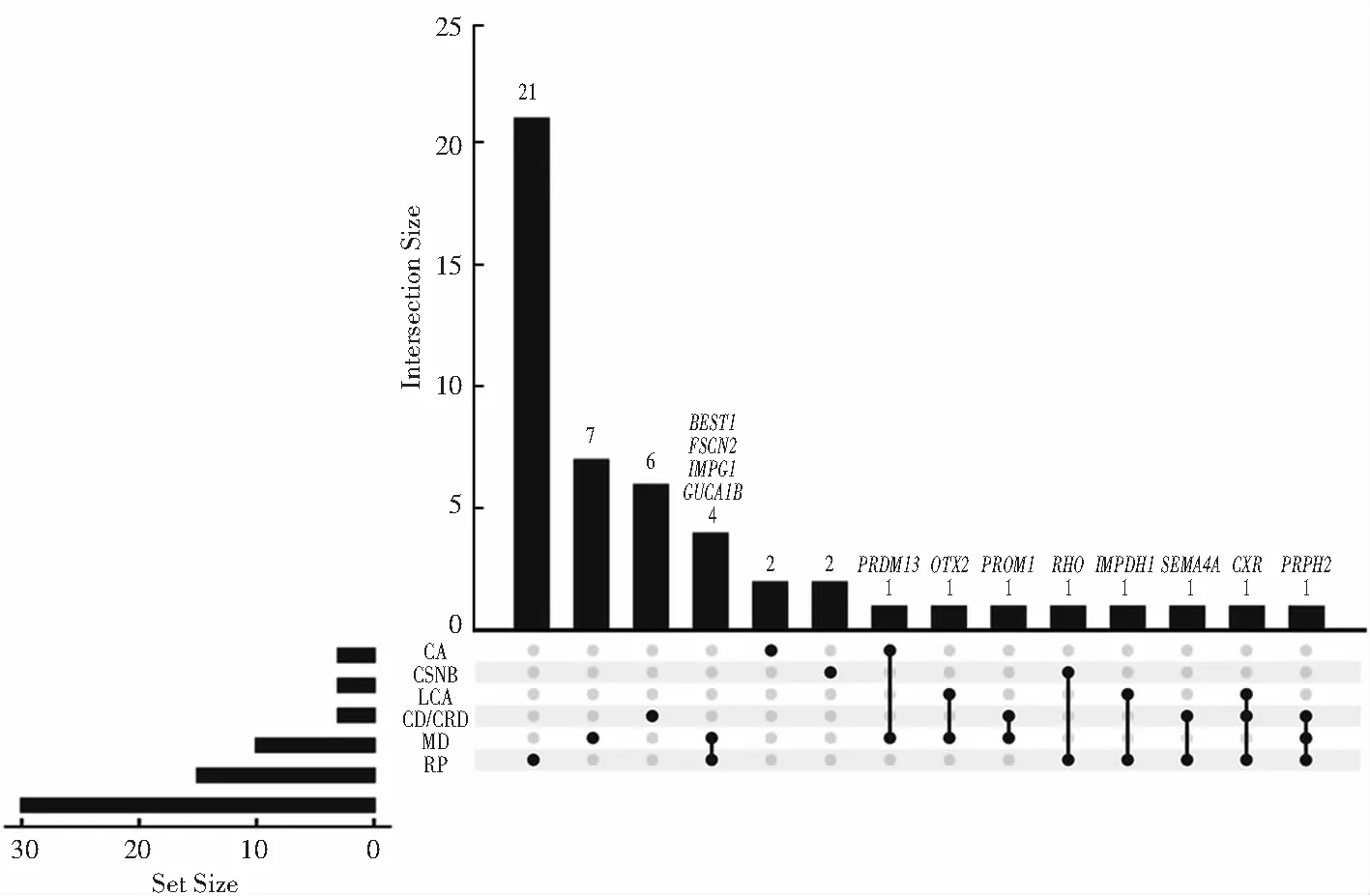
图1 非综合性常染色体显性IRDs相同致病基因统计图 同时引起MD和RP的基因有BEST1、FSCN2、IMPG1和GUCA1B;同时引起CA和MD的基因有PRDM13;同时引起LCA与MD的基因有OTX2;同时引起CD/CRD与MD的基因有PROM1;同时引起CSNB与RP的基因有RHO;同时引起LCA、CD/CRD及RP的基因有CXR;同时引起CD/CRD、MD及RP的基因有PRPH2 CA:脉络膜视网膜萎缩;CSNB:先天性静止性夜盲;LCA:先天性黑矇;CD/CRD:视锥或视锥-视杆营养不良;MD:黄斑变性;RP:视网膜色素变性
3 常染色体显性IRDs的发病机制
常染色体显性IRDs的发病机制主要分为3种形式[15]:(1)功能缺失,即使基因产物失去功能的突变。致病性的产生是因为1个等位基因发生突变失去正常功能,而另1个等位基因不足以维持正常功能,这种情况被称为单倍不足性,通常这种类型的突变会引起常染色体隐性遗传疾病[16]。由单倍不足性所致的显性突变较少见,例如引起常染色体显性遗传RP(autosomal dominant RP,adRP)的前mRNA加工因子31同源基因PRPF31的大多数突变[17]。(2)功能获得,即突变的蛋白与原来正常的蛋白相比较,产生了一种新的功能,但是这种功能并非机体所需,并且会干扰正常的生理途径,造成机体进入病理状态,从而导致疾病的发生。这种情况在常染色体显性遗传病中比较常见。例如RHO基因突变,RHO可以编码与光转导有关的光敏视紫红质蛋白,由于突变的视紫红质分子在光感受器中高水平表达,导致一些细胞机制,例如运输或蛋白降解等无法发挥正常功能[18]。(3)显性-负性效应,即由于1个等位基因突变后产生的异常蛋白分子与正常等位基因产生的正常蛋白分子竞争同一个受体作用位点,竞争性抑制了正常蛋白生理功能的发挥。这种情况下,正常蛋白表达水平越高,越不容易发病;反之,正常蛋白表达水平越低,越容易发病[19]。例如PRPF31基因突变患者中,其发病机制除功能缺失外,在错义突变等情况下还存在显性-负性效应[20]。
CRX是光感受器发育和维持的关键基因,调节多种视网膜基因的转录,研究表明CRX突变可以引起CRD 2型或LCA 7型,多数情况下致病机制为显性-负性效应或功能获得性[21],CRX突变的患者仍然具有1个正常拷贝,但突变的蛋白可能会对正常的蛋白产生干扰,因此添加更多的正常基因是否能使视网膜恢复功能仍属未知。在这种情况下,为了证明基因增强策略是否具有可行性,研究者采用患者诱导多功能干细胞,构建了显性CRX-LCA的视网膜类器官模型,并使用腺相关病毒(adeno associated virus,AAV)介导的CRX基因增强治疗,最后恢复了2个患者来源类器官的部分CRX蛋白功能,证明了基因增强策略对于常染色体显性遗传LCA具有治疗潜力[22]。
4 常染色体显性IRDs的临床特征
常染色体显性IRDs临床表现非常复杂,根据显性性状的表现特点,主要分为完全显性遗传、不完全显性遗传、不规则显性遗传、共显性遗传和延迟显性遗传5种类型[13],但大多数情况下表现为完全显性。(1)完全显性 指杂合子表现出与显性纯合子完全相同的表型。如果双亲之一是患者,子女中发病的概率为1/2。若双亲都是患者,子女中发病的概率为3/4。若患者为致病基因的纯合体,则子女全部发病。此病通常与性别无关,男女发病机会均等。(2)不完全显性 指杂合子的表型介于纯合显性和正常的纯合隐性之间。不完全显性遗传中杂合子全部发病,但病情轻于纯合显性突变患者,显性基因不能完全遮盖隐性基因的作用,即2个等位基因中,1个突变基因不能完全遮盖另1个正常基因的作用,导致杂合个体表现为双亲性状的中间类型。如在植物中,红色菊花与白色菊花杂交后会表现为粉色菊花[23]。(3)不规则显性 由于修饰基因或环境因素等的影响使显性基因杂合体的预期临床性状未表现出来,造成疾病的外显率降低,也就是临床中常见的不完全外显现象,可在家系谱中出现隔代遗传,如视网膜母细胞瘤(图2)。不规则显性的另一特点是临床表现度不同。表现度的差异是指同一种致病基因突变的临床表现程度可有轻、中、重度的不同。来自同一家系、具有相同显性致病基因突变的不同家族成员有时疾病的严重程度并不相同,就是由于表现度差异造成的;一些本应发病的患者可以成为临床表型正常的致病基因携带者,而他们的子女仍有1/2的概率发病,出现隔代遗传。修饰基因是造成表现度差异的原因之一,对某种遗传性状并无直接影响,但可以加强或减弱与该遗传性状有关的主要基因的作用[24]。如果本身具有同一表型效应则与累加基因没有区别。除了修饰基因的不同,环境因素在发育和成长过程中的不同也可以造成一些疾病的症状不同,病情进展速度不同。(4)共显性 指1对等位基因之间没有显性和隐性的区别,在杂合状态下,2种基因的作用和表型同时完全表现[13]。例如,纯种白毛马和纯种红毛马的后代可以同时有红毛和白毛。(5)延迟显性 指某些带有显性基因,如显性致病基因的杂合体,并非在出生后即表现出相应的症状和体征,而是发育到一定年龄时,该基因的作用才表达出来[25],如延迟性家族性耳聋。

图2 视网膜母细胞瘤一家系[24] Ⅲ1-7兄弟姐妹7人中4人发病,约占兄弟姐妹的一半,其亲代Ⅱ2发病,符合典型常染色体显性遗传的发病规律;在Ⅱ2的同胞4人中只有1人发病(1/4);但在Ⅲ代中的Ⅲ13是先证者(同代中首先发病的个体),其4个同胞中虽有2人发病,但父母表型正常,这种情况下,Ⅱ6通常是由其父(Ⅰ2)得到了致病基因的杂合子,由于环境和遗传上各种因素的作用而未能外显,却将致病基因传给Ⅲ12和Ⅲ13,表现为隔代遗传 □:健康男性;○:健康女性;■:男性患者;●:女性患者;:先证者
5 常染色体显性IRDs的基因治疗
对于显性IRDs,可以根据相应的发病机制采取不同的基因治疗方法[17](图3)。(1)功能缺失性疾病,通常选择具有高转换效率和优良安全性的AAV相关载体或非病毒载体转入正常基因,从而恢复表型[26]。(2)功能获得性疾病,由于其突变蛋白干扰正常的生理途径,造成机体的病理状态,需要通过基因编辑的方法,纠正其突变基因位点,使之只表达正常的蛋白,不再表达由突变等位基因产生的有害蛋白,进而消除造成疾病的根源。(3)显性-负性效应类疾病,目前研究发现可以通过基因增强疗法转入正常基因[27-28],使之表达更多的正常蛋白,竞争性抑制异常蛋白与同一蛋白受体的结合,使更多的正常蛋白能履行其生理功能。然而需要注意的是这种方法可能只会减轻症状,为了治愈这类疾病,将来在基因编辑技术允许时,需要在正常基因补充的同时,在RNA或DNA水平上纠正突变等位基因[29-30]。其中包括3种策略:①双等位基因失效并转入正常基因 其原理是设计同时靶向突变型和野生型等位基因的分子,从而在提供外源基因拷贝之前阻断蛋白质的产生;②使突变等位基因失效 在保留野生型的同时,特异性靶向突变等位基因,使之不能表达有害蛋白;③修正突变的等位基因 这种方法将基因组编辑技术与同源重组偶联,在DNA水平上特异性地纠正突变,也就是用正常的序列替换突变部分。

图3 显性IRDs基因治疗策略流程图[17] 功能缺失突变目前可以通过基因增强疗法来治疗;显性-负性效应突变目前可以通过基因编辑疗法或者基因增强疗法来治疗;功能获得突变可以利用基因编辑疗法或基因静默疗法与基因增强疗法相结合的方式来治疗
在IRDs中,RP为常见的临床类型,目前已鉴定出约有30种基因可以引起adRP(表1)。研究指出,与前体RNA剪接体蛋白编码相关的基因(剪切基因)能够引起adRP[31]。目前已被证实能够引起adRP的剪切基因有PRPF31、PRPF3、PRPF6、PRPF8、RP9和SNRNP200。值得关注的是,上述6个剪切基因所编码的蛋白在生物体各种细胞内广泛表达,目前研究发现这6个基因相关的突变只能引起视网膜疾病的发生。因此,针对剪切基因与RP发病的相关研究显得尤为必要。这些基因编码的蛋白质对人体所有类型细胞的剪接功能都是必不可少的,但这些突变在人体其他器官并未造成病理改变,只在视网膜视杆细胞上造成了病理改变。可能因为视细胞存在持续不断的外节膜盘的脱落和更新,对相应突变造成的剪切异常更敏感。PRPF31基因突变是adRP中常见的原因之一,占所有adRP病例的10%[32-33],是导致视网膜色素上皮细胞和视细胞功能缺陷的重要因素。
针对PRPF31基因错义突变和PRPF31基因大片段缺失导致的半合子突变的功能研究结果表明,突变的最终整体效果是导致细胞核中具有正常功能的蛋白水平降低。PRPF31基因的显性突变只在视细胞中才会显露出剪接功能的不足,因为其外节随着膜盘的持续脱落需要持续不断地及时更新,所以对剪切功能有非常高的需求,需要较多的正常PRPF31蛋白。对于人体而言,PRPF31基因虽然存在于很多细胞内,但PRPF31基因的显性突变只影响了视细胞,而对其他细胞都没有造成病变;这个结果表明,1个正常等位基因产生的具有正常功能的蛋白质总量,足以满足除视细胞以外其他所有普通细胞内的基本剪接需求,从而维持正常的细胞功能[34-35]。尽管存在多种机制调控RPPF31基因的表达,蛋白的表达量依旧是临床表现发展的决定性因素。因此,理论上有2种合适的基因治疗方法,一是转入正常基因的基因增强疗法[36],二是通过对突变等位基因进行特异性纠正的基因组编辑疗法[37]。
对于隐性遗传病,一般用正常基因转入病变细胞的基因增强疗法进行治疗,例如IRDs的第1种基因治疗药物Luxturna®,带有RPE65基因的AAV2/2载体,用于治疗RPE65相关的RP或LCA 2型[38],目前已进入市场。近期相关研究者用AAV转入正常PRPF31基因,这种用于PRPF31基因显性突变的基因增强疗法如果获得成功,则不但开启了用于治疗常染色体显性遗传病的一种实用的方法,对于验证显性-负性效应在PRPF31突变病理机制中的作用也有重大意义,未来将对这类显性IRDs的基因治疗带来示范性作用。
总之,虽然目前IRDs仍是无法治愈的疾病,但随着基因治疗的快速发展,有望延缓IRDs的发生并恢复患者的部分视力。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
智慧健康(2021年17期)2021-07-30
国际检验医学杂志(2021年7期)2021-04-15
中学生理科应试(2019年4期)2019-07-08
中国生殖健康(2019年3期)2019-02-01
消费导刊(2017年24期)2018-01-31
辽宁大学学报(哲学社会科学版)(2017年3期)2017-06-21
现代检验医学杂志(2016年5期)2016-08-20
法医学杂志(2015年4期)2016-01-06
中学语文(2015年27期)2015-03-01
继续教育研究(2014年2期)2014-02-27
