
DNA信息存储中关键生化方法的研究
2021-07-21 09:30郜艳敏唐梦童刘倩乔宏艳王桃雪齐浩
合成生物学 2021年3期
郜艳敏,唐梦童,刘倩,乔宏艳,王桃雪,齐浩
(天津大学化工学院,系统生物工程教育部重点实验室, 天津 300350)
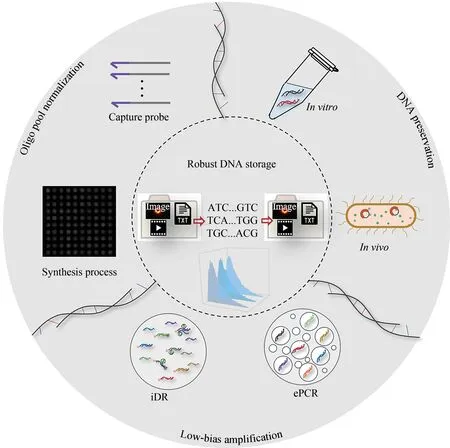
与传统存储介质相比,DNA凭借保存时间长[1-2]、存储密度高[3-4]、易复制等优势,为大规模的数据备份提供了可能[5-7],并有极大的潜力成为新一代存储和检索数据的介质[8-9]。DNA信息存储的发展已有50多年的历史,发展史如图1所示[10-27]。早在20世纪60年代,由计算机科学家Wiener[10]和Neiman[11]首次引入了基于DNA数据存储的概念“基因记忆”。1995年,普林斯顿大学教授Baum正式提出构建基于DNA分子的大容量数据库存储体系[12]。随后在1996年,DNA数据存储的概念首次被Davis的“Microvenus”进行实验验证[13]。他将35 bit的黑白图标“写入”18 bp的DNA序列(CCCCCCAACGCGCGCGCT)并成功解码。1999年,Clelland及其团队[14]开发了一种DNA隐写技术,该方法再次证明了DNA数据存储的概念,并且它是第一个而且是直到2012年唯一一个在体外实现DNA数据存储及恢复的方法。2001—2010年,体内DNA信息在信息存储容量及编码方式上都有了极大提升[15-19]。随着DNA合成和测序技术的发展,2012年,哈佛大学的Church等[20]将一本图书(650 KB)存储在DNA中,2013年Goldman及其同事[21]在DNA中实现了720 KB数据的高容量存储。随后的2015年和2016年,Grass等和Blawat等在合成的DNA中分别实现了0.08 MB和22 MB数据的高容量存储并进行了无错误的检索[22-23],这是DNA信息存储领域的另一个里程碑。2017年,Erlich和其同事[24]开发了一种高效可靠的DNA存储策略——“DNA喷泉”(DNA fountain),利用这种编码机制,可以最大化DNA的数据存储能力。同年,Shipman等[25]利用CRISPR-Cas系统将一张黑白图像和一部短的视频文件“写入”大肠杆菌的基因组中。2018年,Organick等[26]将超过200 MB的数据“写入”DNA中。2020年,天津大学Qi等[27]利用混菌培养系统将445 KB的数据存入细菌体内。一系列的研究表明DNA应用于信息存储具有巨大的发展潜力,而进一步探索这一新型的数据存储体系,对大数据时代海量数据信息的长期存储具有重大的意义。
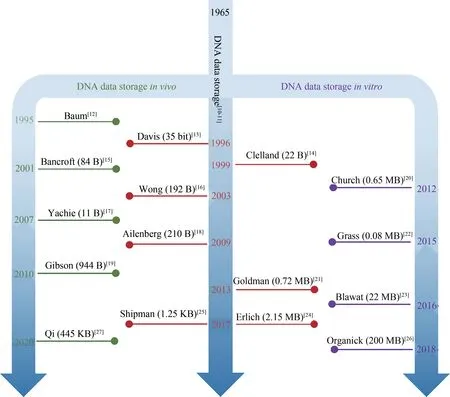
图1 DNA信息存储发展史[10-27]Fig.1 History of DNA data storage[10-27]
DNA数据存储的一般流程是:数字信息编码为DNA序列(编码)—编码信息写入DNA分子(合成)—选择合适的载体将合成的DNA序列进行保存(存储)—利用特定的引物进行有选择地访问(检索)—读取分子(测序)—根据解码规则将DNA序列中的信息复原(解码)[28-30],如图2所示。然而,据报道,整个DNA数据存储流程中涉及的包括合成工艺、保存方法(液体、干粉、包封等)和扩增方式等多个生化反应均会造成序列丢失,增大序列间的不均衡以及碱基突变(替换、插入和删除)等,进而影响DNA数据存储的应用,如图3所示。Chen等[31]基于数百万条序列的不均一性问题,发现造成DNA序列偏差的两个最重要的来源是合成和扩增过程。Grass等[32]发现DNA分子内的错误主要由合成和测序造成,而DNA序列的丢失主要是由保存条件不当引起。Erlich等[24]进行了连续10次的PCR反应,发现第10次富集之后,覆盖度的分布峰更加偏斜,而且要实现完美解码所需要的测序数据量是第1次的6.7倍,说明PCR过程增加了DNA序列的不均一性。而基于目前所采用的解码策略,在DNA序列的拷贝数差异过大的情况下,要想实现成功解码,所需的测序深度较深,造成测序成本增加;丢失超过所能容忍丢失的DNA序列数势必造成信息的丢失,无法实现完美解码;虽然用纠删码可以解决碱基的替换、插入和删除中的部分错误,但势必会造成测序资源的浪费、计算量的增加以及解码时间的增加[31-32]。
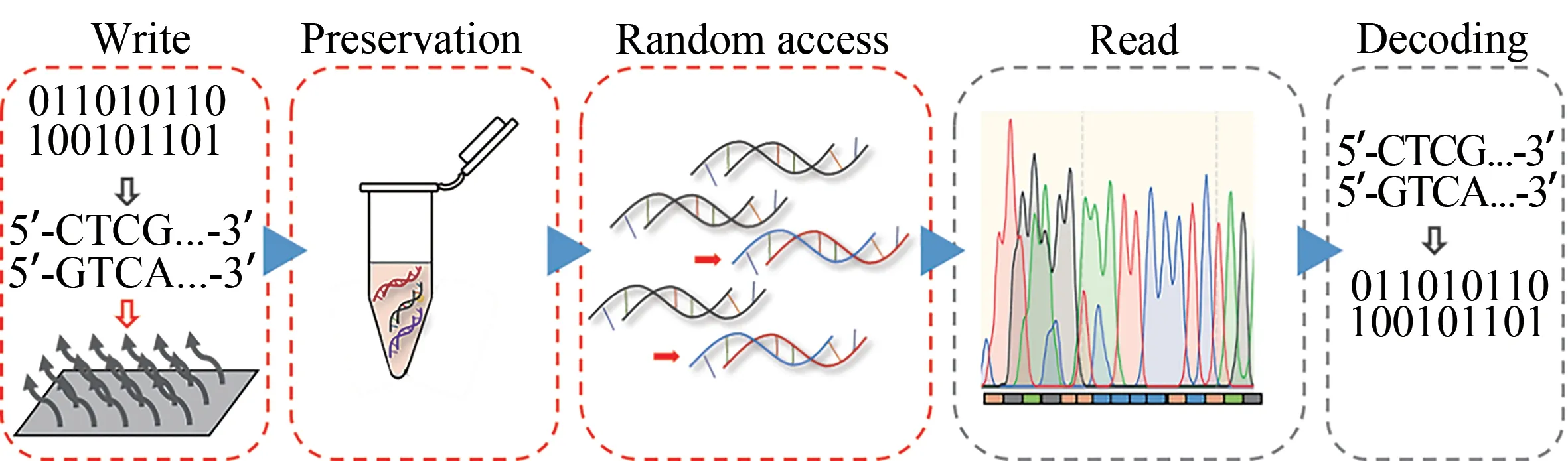
图2 DNA信息存储流程图Fig.2 Flowchart of DNA data storage
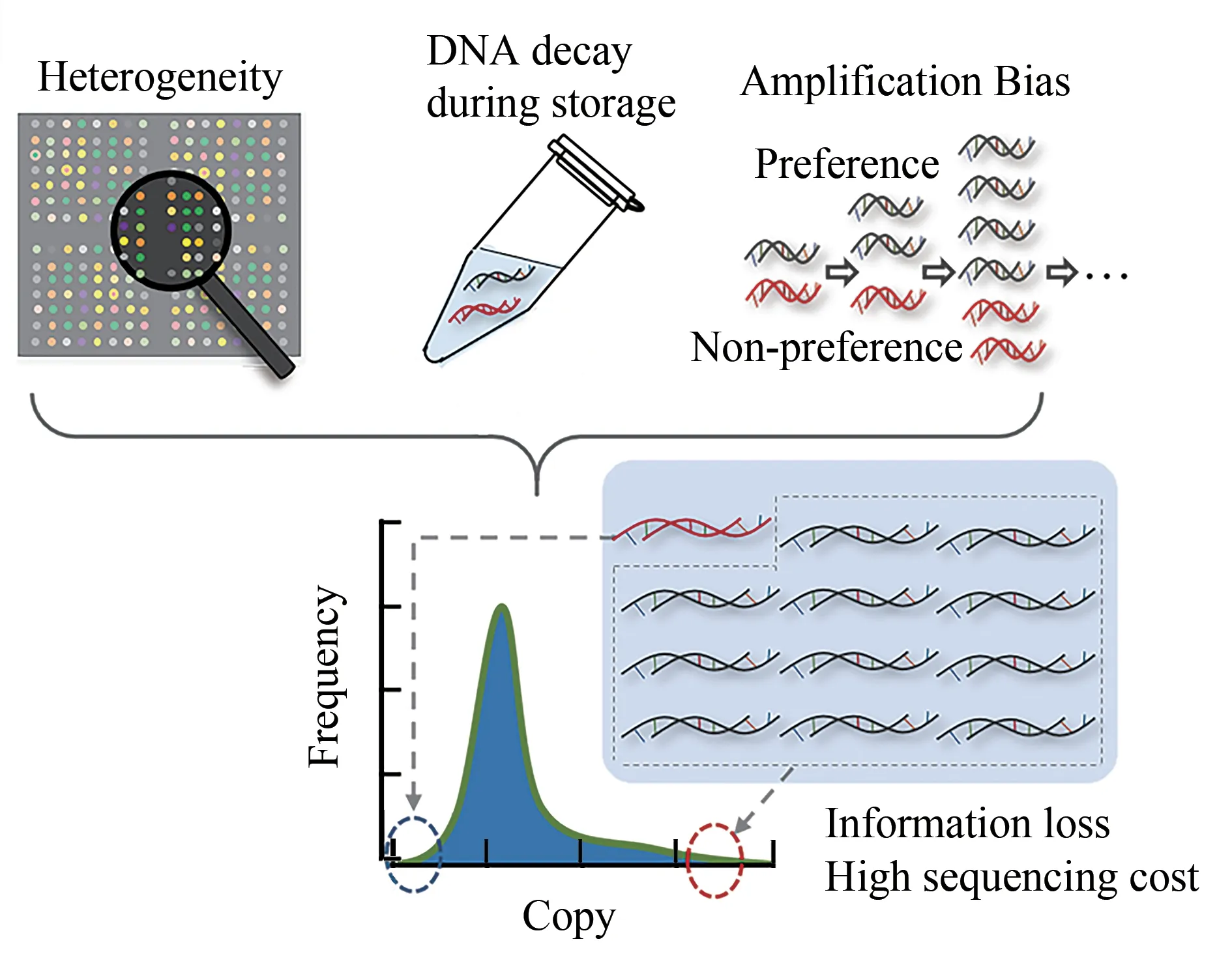
图3 DNA信息存储过程中出现的生化问题Fig.3 Biochemical problems in DNA data storage
为解决这些问题,科学家们提出了各种各样的解决方案。本文以DNA信息存储为主线,详细介绍了DNA数据存储过程中的一系列生化反应对携带有信息的大规模寡核苷酸文库造成的影响,重点介绍了现阶段为解决DNA信息存储中的这些问题所采取的DNA分子合成、保存以及扩增方法,论证了这些生化方法对操纵大规模寡核苷酸文库的可行性和有效性,最后总结并讨论了目前该领域所涉及的生化反应存在以及亟需解决的主要问题。
1 合成和均一化方法的优化以实现寡核苷酸文库的均衡性
目前DNA合成的方法主要有:芯片合成、柱式合成以及酶促合成[8,33-34]。芯片合成具有可合成任意序列、通量高、成本相对较低等优势[35];柱式合成具有可合成任意序列、准确性高等优势,但其通量较低[36];酶促合成[37-39]具有潜在的低成本、高保真、高效率等优势,最近十几年备受关注,然而由于技术尚未成熟,目前还未进入大规模应用阶段。而目前应用于DNA数据存储的寡核苷酸文库需要数万条甚至数百万条序列,现在能够满足此要求的只有芯片合成技术。芯片合成过程中产生的同一种DNA序列均达数百万个拷贝,但此过程中可能会发生碱基的替换、插入和删除等错误,造成同一序列的每个拷贝出现各种不同的错误[40]。而且受目前芯片合成技术的限制,每个DNA序列的合成总量与其在芯片上的空间位置有关,分布在边缘部位相比其他部位的DNA序列合成量较少,这将造成寡核苷酸库中DNA序列具有很大的不均一性(单链DNA分子分布的不平衡性也即各个序列的拷贝数具有很大的差异性)[31,41]。另外,不同的芯片合成技术合成的寡核苷酸的质量也有很大的差异,并且合成质量与合成成本之间是成正比的,那么以较低的合成成本实现DNA数据存储是科学家们所追寻的。
1.1 合成工艺改进
2019年,微软研究院联合Twist Bioscience将每个序列的测序结果映射回其在合成芯片上的位置,结果表明DNA序列的合成偏差与其在芯片上的空间位置有关[31]。为了解决空间位置对合成质量的影响,Twist Bioscience对单体核苷亚磷酰胺进行专有的化学修饰增加合成工艺的耐受范围,同时还对化学工艺参数进行了优化,确保在较短的时间内使流通池中的化学试剂更加均匀地分散。合成工艺的改进使得芯片上的每条寡核苷酸的数量更加均匀,其分布已呈现了较好的正态分布,而且这一改进使得合成的错误明显降低。
另外,近些年酶促合成方法也取得了一些研究进展。2019年,Lee等[38]利用末端脱氧核苷酸转 移 酶 (terminal deoxynucleotide transferase,TdT)开发了从头酶促合成策略,并依据该酶的酶促合成性质设计一种特殊的编解码方法应用于数据信息的存储。但因其合成准确度较低且通量不高,不能用于大规模的数据存储,因此该方法的应用受到了一定的限制。2020年,Antkowiak等[42]开发了一种依赖于大规模平行合成的DNA存储系统,该方法的成本远远低于传统的柱式合成方法,但提高合成速度时,其序列错误率随之增加。同时Tabatabaei等[43]采用传统的穿孔打卡记录数据原理,将酶(Pyrococcus furiosusArgonaute)作为“打孔器”在现有的双链DNA上留下“刻痕”,出现“刻痕”表示1,没有则表示0,进而存储数据。该方法完全不涉及合成,也不会出现合成过程中出现的一些问题,但其存储容量和密度都较低,这损失了原本DNA信息存储的优势。
1.2 构建均一化的寡核苷酸文库
根据目前固相合成机制可知,基于芯片合成寡核苷酸文库的合成是不完美的。在核苷酸的添加过程中,有可能添加意外终止造成寡核苷酸合成不完全,或者一个核苷酸没有添加上造成DNA序列上碱基的删除[44]。2018年,Zhang等[45]开发了一种化学计量标准化的寡核苷酸纯化(stoichiometrically normalizing oligonucleotide purification,SNOP)方法(如图4左),该方法可以同时纯化并均一化数百种不同的寡核苷酸。其作用原理是:对于每个寡核苷酸Oi,设计一个相应的前体寡核苷酸Pi。该前体包含了标签序列和寡核苷酸Oi,标签序列区位于前体DNA的5′区,从5′到3′依次是所有前体共有的通用序列、特异性的条形码序列和脱氧尿嘧啶(dU)核苷酸,其中每个条形码序列均由多个交替的强(G或C)和弱(A或T)核苷酸(如CTCTCT或CAGACT)组成。同时,3′端修饰有生物素基团的捕获探针(单个合成)与对应的前体序列的通用序列区和条形码区互补,以这种方式设计条形码序列的目的是最小化每个寡核苷酸与捕获探针杂交的标准化自由能的变化,使得每种前体都最有利地与其对应探针完美结合。在SNOP过程中,等摩尔比混合每个捕获探针,当捕获探针是限制性试剂时,尽管初始前体浓度不同,但每种全长前体的杂交量相似。随后使用链霉亲和素包被的磁珠进行固相分离以除去未结合的前体,然后使用USER enzyme mix从dU位点上裂解得到寡核苷酸Oi。除了可以提高寡核苷酸的纯度,SNOP方法还可以对寡核苷酸的浓度进行均一化处理,以便使最终的寡核苷酸文库中的每条寡核苷酸的浓度相似。作者通过对含有64条和256条寡核苷酸的寡核苷酸文库进行均一化实验验证了该方法的有效性,测序结果表明即使前体Pi浓度存在很大差异的情况下,得到的产物Oi的浓度也相似。这是到目前为止首个报道的对寡核苷酸文库纯化及浓度标准化的方法,该方法为后续进一步开发寡核苷酸文库均一化方法提供了方向,但该方法需要前体DNA序列和需要化学修饰。
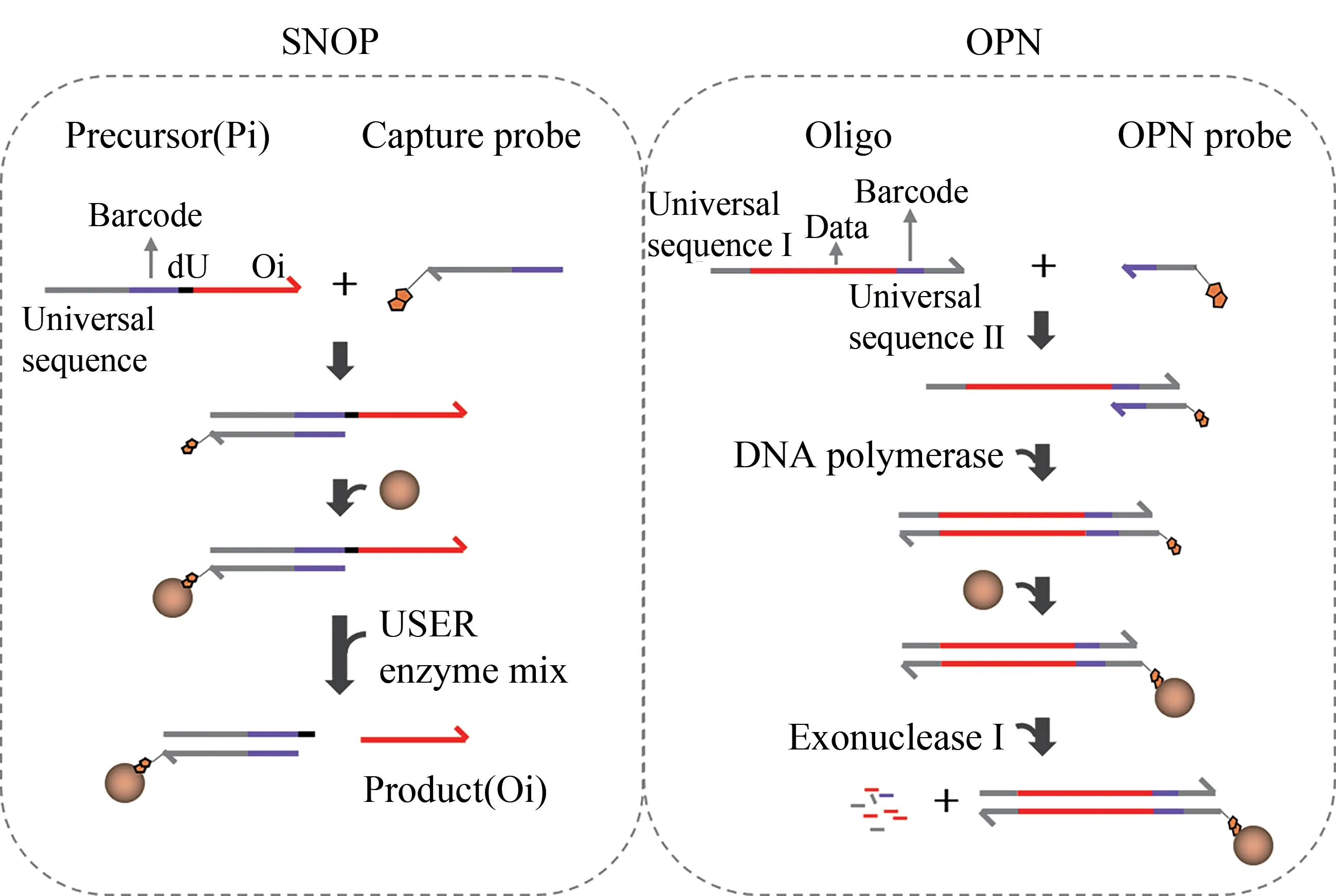
图4 两种大型寡核苷酸文库DNA的均一化方法Fig.4 Two different DNA normalization methods of large-scale oligo pool are shown:SNOPand OPN
在2020年,Gao等[46]对该方法做了进一步的改善以降低其成本,改进的方法被称为寡核苷酸文库均一化(oligo pool normalizing,OPN)(如图4右)。这里,寡核苷酸序列被设计为引物序列、中间可变序列区域以及标签序列区域(5′→3′),该标签序列区域包括条形码区域和通用序列区(5′→3′)。同时,捕获探针的5′端修饰有生物素并与标签序列区的序列互补(每个捕获探针分别合成)。在OPN过程中,首先使用正向引物和磷酸化的反向引物进行寡核苷酸的扩增,然后用Lambda外切酶进行降解以富集寡核苷酸文库。等摩尔比混合每个捕获探针,当捕获探针是限制性试剂时,尽管每个初始的寡核苷酸浓度不同,但每种寡核苷酸的杂交量相似。随后加入DNA聚合酶沿杂交到寡核苷酸上的捕获探针的3′端进行延伸,使其修补成双链。之后加入外切酶Ⅰ将未结合的寡核苷酸进行降解,最后使用链霉亲和素包被的磁珠分离,得到相对标准化的寡核苷酸文库。首先使用256条寡核苷酸文库进行了验证。后进一步对OPN方法进行了改善,构建了OPN2.0。在以下几个方面做了改进:①使用生物素化的正向引物和磷酸化的反向引物进行寡核苷酸文库扩增,这样使得捕获探针无需再进行任何的化学修饰,从而降低成本;②通过改变通用序列区序列,在一个寡核苷酸文库中使用了4个不同的通用序列,组合条形码区域,寡核苷酸文库扩大至1024条,这是目前报道的均一化的最大的寡核苷酸文库。这也从技术上进一步说明,通过将一组精心设计的通用序列与256个条形码组合,理论上可以同时均一化300万条寡核苷酸序列。
改进合成工艺可能是实现寡核苷酸文库均衡性最本质的解决方案,但其势必会造成合成成本的大幅度增加。寡核苷酸均一化方法是比较可行的解决方案,在没有较大地增加合成成本的基础上实现寡核苷酸序列的均衡性,进而降低测序成本,实现完美解码。
2 保存方法的优化以实现寡核苷酸文库稳定性的存储
DNA数据存储最重要的优势之一是其保存时间较久(长达几个世纪)。然而,与传统的基于磁性或光学的存储方式相比,DNA数据存储的稳定性仍是一个重要的问题。DNA在较恶劣(如高湿和紫外线)及温度较高的情况下,很容易被烷基化、水解及氧化[47-48],进而造成DNA序列丢失,碱基的替换、插入和删除,从而造成数据信息的丢失[32,49-50]。因此,实现DNA的长期稳定保存对于DNA数据存储至关重要[23]。目前,用来实现DNA保存的方法主要有液状、干粉、封装、DNA与碱性盐混合干燥、非天然核酸和体内存储6种形式(图5)。
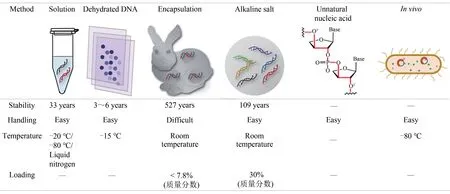
图5 6种不同DNA保存方法Fig.5 Six different storage methods
2.1 液体法
在实验室中长期保存DNA的传统方法是以液状形式保存DNA[51]。DNA在酸性条件下易水解,所以常将DNA溶于TE缓冲液中,并将其置于低温冰箱(-20℃或-80℃)或液氮中保存[52]。但此种方法需要的能耗大,这对于大规模的数据信息存储是不可取的。
2.2 粉末法
DNA粉末保存也是长期储存DNA的一种实用方法[53-54]。固态DNA的降解主要受大气中的水和氧气的影响,所以将干燥的DNA样品保存在相对较低湿度下是非常重要的[49,55-56]。脱水会降低DNA分子的流动性,并抑制DNA的脱嘌呤、脱嘧啶、脱氨和水解反应。另外,据报道,海藻糖对DNA二级结构具有很强的稳定作用,在存在海藻糖的情况下,固态的天然DNA即使加热到120℃也不会变性。这种稳定作用可能是海藻糖与磷酸盐结合中和DNA的负电荷或者是海藻糖与DNA之间的氢键建立的网络减少了DNA结构的波动(玻璃化假设)[55-57]。制备干燥DNA的方法有以下几种:喷雾干燥、喷雾冷冻干燥、空气干燥以及冷冻干燥,其中冷冻干燥是成本最低、最受欢迎的方法[58]。将干燥的DNA通过特定的方式固定在纸或玻璃板上是在室温下存储的另一种选择,2019年,Newman等[59]将DNA粉末固定在玻璃板上,并通过数字微流控设备(digital microfluidics,DMF)实现DNA的随机提取。这种方法不仅可以实现大量数据物理上的隔离,而且能够实现数据信息的随机检索,对于大规模的数据存储是非常重要的。另外可以加入一些商业化的DNA稳定剂如DNAStable,与DNA共干燥实现核酸在室温下的长期保存(50℃下300多天)[60-61]。
2.3 封装法
通过封装将DNA与外界环境隔绝开来,可避免环境变化(如高温等)造成的DNA损伤[62]。Grass等[22]和Paunescu等[63]提出利用化学稳定性和热稳定性较好的二氧化硅对DNA进行封装,使得DNA在60℃可保存2个月(相当于在室温可保存2年),但DNA载量很小。Koch等[64]将封装有DNA的二氧化硅融合到3D打印材料和眼镜制剂中,实现了嵌入信息型物质的制备。Chen等[65]通过应用具有交替的DNA层和聚阳离子分子[即聚乙烯亚胺(PEI)]的逐层(LbL)设计,将DNA结合到磁性纳米颗粒上,同时保护性二氧化硅层生长在多层纳米颗粒的顶部,以保护DNA免受外部损害。该方法将DNA载量提高到7.8%(质量分数)并且使得DNA在室温下可保存20~90年(10℃下150 bp长度的DNA可保存527年)。
2.4 碱性盐混合法
Del Valle等[66]已经证明吸附到羟基磷灰石上的DNA可以免受DNase I的降解。2020年,Kohll等[67]利用磷酸钙、氯化钙和氯化镁等碱金属与DNA混合干燥,将DNA包裹在这些碱金属盐中,将DNA载量提高到30%(质量分数),同时可实现DNA的长期保存(10℃下可保存109年)。相对于二氧化硅DNA包封法,该方法载量高、易操作,但其保存时间没有包封法长。
2.5 非天然核酸法
α-L-呋喃糖基核酸(α-L-threofuranosyl nucleic acid,TNA)是一种非天然核酸,由2′,3′-磷酸二酯键连接带有碱基的四碳糖而成[68]。其中,2′,3′-磷酸二酯键不容易被核酸酶酶解。Yang等[69]将DNA上带有的数字信息通过碱基互补配对转移到TNA中,成功抵御了核酸酶的酶解,可防止核酸酶酶解导致的信息丢失,但目前其稳定性还没得到验证。
2.6 体内存储
除了体外存储,DNA体内存储也有很大的优势,如低成本复制和长久稳定保存[70-73]。从DNA信息存储发展的早期到2012年,DNA信息存储全部都是在体内进行[15-18]。2010年,Gibson等[19]首次将人工合成的一个支原体基因组(1 077 947 bp)存入酵母细胞中并成功进行复制和传代,这在将体外信息存储在细胞中的历史上具有里程碑意义。2017年,哈佛大学的Shipman等[25]通过CRISPR-Cas编辑工具将一部无声短片(2.6 KB)存入细菌体内,并实现了90%数据的恢复。2020年,Hao等[27]利用同源重组技术将携带有445 KB的数据信息的DNA序列文库组装到高拷贝质粒中,并转化到细菌细胞进行混合培养,同时进行了5次传代,数据恢复率达到98%以上,这是目前报道的数据量最大的体内存储。但体内存储也存在一定的缺点,其存储密度相比体外存储低,而且DNA在生物体内会发生损伤,可能导致碱基的替换、插入或删除等。另外,虽然细菌细胞可以存在数百万年,但其携带数据信息的稳定性没有明确的报道。
3 扩增反应的优化以实现低偏好性地放大寡核苷酸文库
DNA数据信息很容易被复制,这也是DNA数据存储得到高度关注的原因之一,文献中报道最多就是利用PCR扩增技术。模板序列的长度、DNA序列以及二级结构、聚合酶的种类、是否有添加剂以及PCR反应条件等均会影响PCR的扩增效率和产物的准确性[74-78]。这对一个具有高度序列复杂性的大型寡核苷酸文库的扩增产生了巨大的挑战。近些年,为解决扩增过程中产生的这些问题,科研工作者展开了一系列的探索。
3.1 优化PCR反应体系
Czerny[79]发现增大引物浓度,可以显著增加扩增产物的产量,说明引物浓度是PCR反应的限制因素;而且,PCR反应过程中过量的引物会在反应的后期阻止非特异产物的生成。因此,对于PCR反应,增大引物浓度不仅可以提高产量而且提高产物质量。Wang等[80]对基于大型寡核苷酸文库的引物进行了寡聚设计,该设计仅一个引物结合位点,将该引物结合位点连接至寡核苷酸的一端以将单链DNA转化为测序所需的双链DNA。同时,他们设计了用于单个引物结合位点的组装原始测序读数中的DNA序列的算法。两种设计的组合不仅可以无错误地恢复超过99%的数据,而且比现有的使用短链DNA信息存储方案的数据存储密度有显著的提高。另外,加入适量的增强剂或添加剂,如DMSO[81]、甲酰胺[82]、甘 油 、 甜 菜 碱[83-84]、 牛 血 清 蛋 白[85]、Triton X-100、乙二醇、核苷酸类似物7-去氮-2′-脱氧鸟苷(dc7GTP)[86]等已经证明了可以改善富含GC的DNA序列扩增,这些小分子增强剂或添加剂既可以阻止模板和引物各自形成复杂的二级结构,也可以增加引物在温度高于溶解温度的情况下与模板结合的机会。
聚合酶是产生偏好性的主要来源,尤其在模板链上聚合开始时。Pan团队[87]使用了包含12个随机碱基的文库以对DNA聚合酶引发的偏好性进行表征。同时使用3′末端带有随机六聚体的引物对合成文库进行扩增。结果表明引物的3′端的6个核苷酸序列以及引物位点下游的4个核苷酸序列会影响引物的引发效率。通过从单引物模板扩增证明了3′端下游的优选引发基序是富含GC的。在65 536条序列中,A家族的DNA聚合酶(Qiagen TopTaq,QTT-A)对序列“GGGGGCGG”具有最高扩增效率,然而B家族的DNA聚合酶(Qiagen HotStar HighFidelity,QHH-B)对该序列的扩增效率仅排在4180名,说明A家族的QTT-A对该序列具有更高的扩增效率。他们将观察到的DNA聚合酶偏好性整合到了引物设计程序上,该程序可指导在模板上设计引物的最佳位置。另外,聚合酶的保真度影响产物的质量(是否有特异性,有无突变),尤其对于长片段DNA(10 kb以上)的扩增,可以考虑使用具有高保真度的DNA聚合酶在较少的扩增循环数下扩增,随后回收产物;然后加入PCR反应组分,再次扩增,如此循环多次。这样在PCR反应进程中可以保证充足的反应组分和高效的酶活,以降低引入突变的概率。
这些研究结果可指导设计PCR引物序列和携带数据信息的寡核苷酸序列、帮助优化PCR反应体系以及为聚合酶的选择提供参考。
3.2 优化PCR反应程序
早在1991年,Don等[88]开发了Touchdown PCR以提高其扩增的特异性,它避免了为确定最佳退火温度而进行的反应条件优化过程。Touchdown PCR是指每隔一个循环退火温度降低1℃或0.5℃,直至降至Touchdown退火温度,并以此退火温度进行10个左右的循环。其原理是较高的退火温度提高引物结合的难度,保证PCR扩增产物的正确性,待靶标DNA序列富集后,再降低退火温度进一步提高扩增效率。但Touchdown PCR最主要的一个缺点是扩增效率较低。随后,Hecker等[89]改进了Touchdown PCR,开发了Stepdown PCR,即退火温度由小幅度的下降和较陡峭的下降组成,这样的改进可以简化热循环仪的编程,同时满足了在复杂模板中提高扩增特异性的需求。在2008年,Frey等[90]又在Touchdown PCR的基础上开发了Slow down PCR,也即通过降低PCR仪的降温速率并在每个温度梯度下进行3个循环来提高引物的退火效率和TaqDNA聚合酶的延伸效率,以实现高GC含量序列的扩增。后来,Aird等[76]证明相比降温速率在6℃/s下扩增的13%~58%GC含量的样品,通过降低PCR程序退火过程中的降温速率(2.2℃/s)可以扩增的样品的GC含量更广(13%~84%)。而且可以通过添加2 mol/L的甜菜碱或者延长变性时间实现高GC含量(23%~90%)的样品的扩增,然而这种会导致较低GC含量的样品没有扩增,使该部分序列丢失。
3.3 e PCR
在常规PCR反应中,多种分子在一个单一的液体体内相互作用。在反应中引入的外源DNA或早期反应步骤中发生的任何错误如碱基错误、引物二聚体或嵌合分子等都可以在整个反应体系中自由地传播而没有任何阻碍。这可能会导致产生非特定或错误的扩增产物。乳液PCR(emulsion PCR,ePCR)[91-92]可通过油包水型乳液将液体分为大量分开的独特反应室(约1010/mL),此时理论上模板DNA的每个分子都被限定在一个独特的反应室中,并随PCR反应的进行而被复制,直到耗尽每个乳液中的所有资源(如图6左所示)。该反应方式避免了常见PCR的缺陷如假阳性、引物二聚体或嵌合体等,同时避免了由于不同DNA序列的扩增效率不同而导致的序列不均衡性,这使得大量DNA序列实现平行扩增而不会造成任何的偏好性[93]。2018年,Organick等[26]将ePCR技术应用到DNA信息存储中,实现了数百万的DNA序列(存储200 MB的数据信息)的同时扩增,并且可以在平均5倍的覆盖度下实现完美解码,这是目前已报道的文献中实现完美解码所需的最少的测序资源。这无疑得益于ePCR技术可尽量减轻由于不同的扩增效率而造成的DNA序列的不均一性,而且避免了原始模板量少而处于扩增劣势的情况下造成的DNA序列丢失。
虽然ePCR有众多优势,但就PCR扩增机制—产物为下一轮的模板而言,这一反应机制会导致序列变异产物不断被扩增,使得错误信息被不断积累[94]。具体而言,若PCR早期发生碱基错误(替换、插入和删除),那么该错误会随PCR扩增呈指数级放大直至反应结束。这将对实现完美解码提出巨大的挑战,也将浪费很大的测序资源,而且它是任何变体PCR都无法解决的问题。并且目前没有很好的方式可使用PCR实现稳定的、重复性的扩增。因此,我们亟需开发新的DNA序列扩增方式来替代目前的PCR技术。
3.4 恒温扩增
恒温扩增技术在近些年得到快速的发展,已广泛应用于生物技术、生物纳米技术以及生物医药等领域。2020年,Gao等[46]开发了一种恒温的DNA读取(isothermal DNA reading,iDR)方式,它在恒温下实现了稳定且可重复的DNA复制(如图6右所示)。具体就是将寡核苷酸文库通过生物素与链霉亲和素的高亲和力结合到磁珠上并联合链置换扩增反应实现数据的可重复性读取,该系统被称之为iDR。使用iDR反应是因为它具有以下几个优点。①其扩增机制是一种线性扩增[95-96],而且只从最原始模板上进行复制,不会将产物作为模板复制。因此,它不会造成更大的DNA序列不均一性,而且完美地避开了碱基错误的扩散,进而节省了测序资源。②该系统实现一次复制之后,可以用磁铁将模板与上清液中产物分离,实现模板的多次重复复制。实验结果证明该系统可以实现至少10次的稳定可重复读取。③该系统可在恒温且室温下进行反应,这为以后的大型数据存储节约了资源。④该系统以可控的方式产生单链或者双链产物,且其产物携带有磷酸基团,为后续反应如构建二代测序文库时加接头提供了便利。该系统结合寡核苷酸文库均一化OPN方法,即使对于合成质量较差的寡核苷酸文库,也可实现寡核苷酸文库的低偏好性、稳定且可重复性扩增。但其扩增效率较低,而且方法不适用于长片段的扩增。
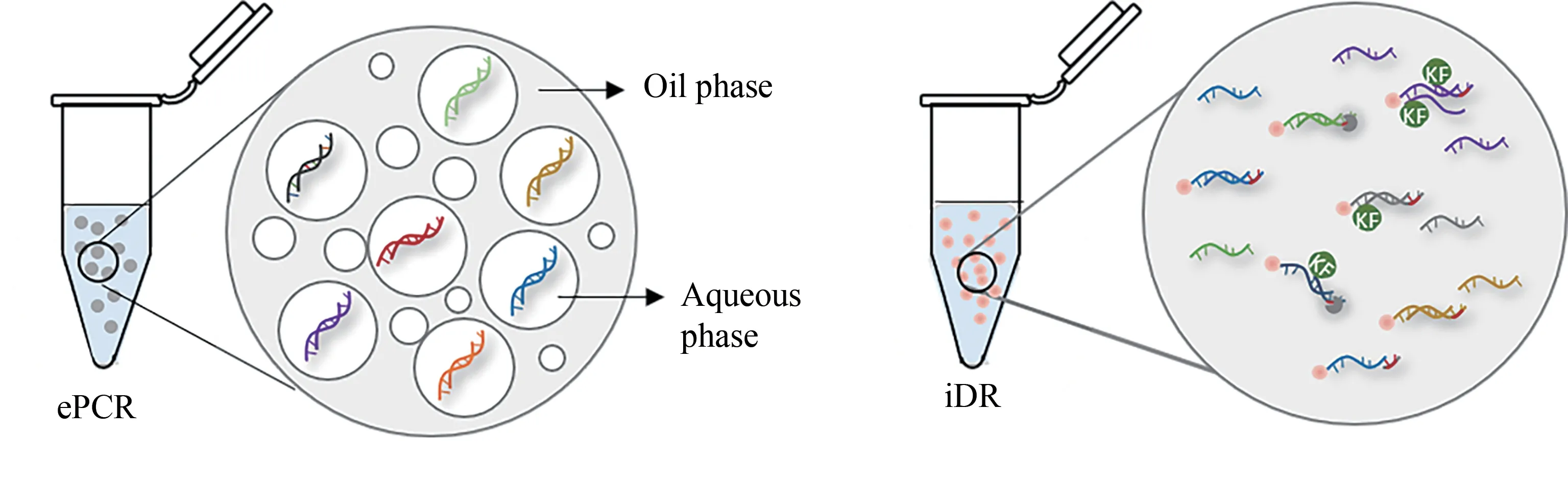
图6 两种大型寡核苷酸文库DNA的扩增方法Fig.6 Two differentamplification methodsof large-scaleoligo pool areshown:ePCR(emulsion PCR)and iDR(isothermal DNAreading)
DNA信息存储过程中,寡核苷酸文库的低偏好性扩增对于数据的完美解码和重复读取非常重要。而目前已存在的扩增方法中,优化反应体系、优化反应程序、使用ePCR以及恒温扩增反应均能降低扩增的偏好性。然而,要实现数据的重复性读取,可考虑以PCR和恒温扩增相结合的方式。
4 结语
随着生物技术的发展,特别是高通量的芯片合成和二代测序技术的不断完善,DNA数据存储领域得到了越来越多的关注。本文对DNA信息存储的发展进行了描述,详细阐述了在该过程中出现的一系列生化问题的原因,针对这些问题提出解决方案,并对其中存在的挑战及问题进行了概括。
首先,芯片合成为寡核苷酸的快速、准确合成提供了有利的保障,伴随着合成工艺的改进,寡核苷酸文库的质量也将大幅度提升,而且寡核苷酸文库的均一性还可以通过均一化方法如SNOP或者OPN技术进一步改善。再者,携带数据信息的寡核苷酸的长期稳定保存关系到信息的稳定性和持久性,通过将寡核苷酸保存在碱性盐中能够模拟类似化石对DNA的保护,可以在较高的DNA载量下实现对核酸的长久保存,虽不及二氧化硅封装DNA对核酸保护的时间久(理论模拟计算保存时间可达数百万年),但其装载量较高(目前报道的装载量最高大于30%,质量分数)且易操作。最后,数据的读取过程需要DNA的复制,由于PCR技术比较成熟、扩增效率较高、存在多种变体PCR且可以利用引物做到随机检索,众多优势使其成为目前使用最广泛使用的方法。但PCR技术也有一些缺点,如产物作模板、扩增偏好性、错误产物的扩散以及产生非目标产物。目前报道的iDR技术由于其扩增机制为线性扩增,可以有效防止文库的不均一性随深度复制而过度放大;序列变异产物不会被复制而防止了错误信息的积累;另外,其产物的5′端携带磷酸基团,非常有利于后续的二代测序过程。但该方法也有一定缺陷:①扩增效率较差;②由于聚合酶缺乏3′→5′外切酶活性,所以产物的点突变的频率增大;③该方法需要长识别序列的缺口酶[因为识别序列越短,编码的难度也会相应的增加(携带信息的片段不能出现该识别位点)],所以可以使用的缺口酶的种类是有限的;④不适用长片段模板的扩增。因此可以将PCR和iDR技术结合,首先利用PCR较高的扩增的效率,使用几轮(<10轮)PCR将商业合成的寡核苷酸文库富集,这样既可以在减少原始文库使用量的情况下得到大量的寡核苷酸文库池,也可以通过其将产物生物素化。然后,生物素化的产物固定在磁珠上,在室温下实现DNA序列稳定、可重复性的扩增。
虽然大量研究表明DNA信息存储无论是在存储能力、保存时间还是稳定可重复的读取上都展现出了巨大的发展前景,但目前DNA信息存储仍面临巨大的挑战。①从大规模应用的角度上看,现阶段的合成和测序成本相对较高,特别是合成费用(约占DNA信息存储的90%)。另外,就目前的合成技术而言,芯片合成序列的长度最长至300 nt,且合成的碱基错误率也急剧增加,合成成本也大幅度提升。②高质量的寡核苷酸文库是DNA信息存储的基石,但由于质量和合成成本是成正比的。目前报道的均一化方法成本相对较高,步骤也相对烦琐。因此,亟需开发新的生化方法对大型的低质量的寡核苷酸文库实施均一化,这样就可以在低合成成本的基础上实现完美的数据存储。③对于大规模DNA信息存储而言,能源消耗也是一个需要考虑的因素。在长时间尺度下实现DNA的稳定保存是非常关键的,而且如何实现数据的物理隔离也是一个亟需解决的问题。④据微软报道,目前一个PCR反应体系可以操纵106种不同的DNA序列,那么体系中并行操纵多少种DNA序列是一个生化反应的极限,为未来生化技术的开发提供了一个方向。⑤DNA序列的重复性读取对于“冷数据”的存储也是非常重要的,当前的文献报道是可以进行20次的重复性读取[97],探索目前的生化技术对于数据重复性读取的极限也是一个值得研究的方向。⑥目前报道的恒温下对寡核苷酸文库的扩增技术受到缺口酶种类的限制,可以利用CRISPR-Cas9突变体作为缺口酶[98-99],可以减少对编码的限制,同时可以做到数据的随机存储。另外,可以优化具有高保真度的具有链置换功能的聚合酶用于恒温反应,产生高质量的扩增产物。⑦基于DNA的生化特性,开发鲁棒的编码策略以及高效的纠删码[100-102]有望弥补现阶段的合成、保存、扩增以及测序技术的不足。
我们期望随着对DNA信息存储和生化技术研究的深入,DNA信息存储领域取得的突破能够使其进入商业应用,并逐步弥补甚至取代当前的数据存储方式。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
湘潮(上半月)(2022年7期)2022-12-06
生物学通报(2022年1期)2022-11-22
中国农学通报(2022年12期)2022-06-01
中国种业(2021年11期)2021-11-25
科学导报(2021年29期)2021-06-03
猪业科学(2021年3期)2021-05-21
中国生殖健康(2020年4期)2021-01-18
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
