
细胞内大片段DNA数据存储的多RS码交织编码
2021-07-21 09:30:56陈为刚葛奇王盼盼韩明哲郭健
合成生物学 2021年3期
陈为刚,葛奇,王盼盼,韩明哲,郭健
(1天津大学微电子学院,天津300072;2教育部合成生物学前沿科学中心,天津大学,天津300072;3天津大学化工学院,天津300072)

人工合成脱氧核糖核酸(DNA)作为一种有潜力的数据存储介质,存储密度高,可用时间久,保存能耗低,有望成为未来海量离线数据存储的重要选择之一[1-7]。美国半导体工业协会(SIA)与半导体研究公司(SRC)在2021年1月发布《半导体十年计划》,将DNA数据存储列为与硬盘、固态硬盘、磁带并列的大量数据的主要存储方式之一,成为未来全球存储产业竞争的重要方向[8]。DNA数据存储的模式主要包括:短片段寡核苷酸池(Oligo pool)存储[9-18]、细胞内DNA存储[19-26]等。短片段的寡核苷酸池存储,借助DNA的高通量芯片合成与测序技术[18],发展迅速,但是在大规模均衡扩增、复制成本方面仍存在很大挑战[12]。细胞内DNA数据存储,尤其是细胞内大片段DNA存储,借助体内组装方法实现短DNA片段组装成长片段,借助体内复制实现高效扩增,复制成本低,在大规模数据分发等场景或有潜在应用价值。近年来,合成生物学发展迅速,尤其酵母基因组的人工合成与基于酵母的同源组装取得了很大进展[27-37]。在此基础上,前期我们设计组装了一条约254 kb的酵母人工染色体,存储了37.8 KB图片与视频数据,除了能可靠复制,未见其他明显生物功能,综合考虑信息编码、合成组装、复制稳定性、采用三代纳米孔测序仪读出等问题,实现了细胞内的外源数字信息写入,并基于三代纳米孔测序器件实现了快速便携读出[19]。目前长基因组的合成与组装难度大、成本高,借助细胞增殖的复制成本低,纳米孔测序可实现便携式快速读出。综合以上几个特点,大片段DNA存储非常类似只读光盘(CD)的早期发展阶段,该种存储模式称为“酵母光盘”或“DNA光盘”模式。本文针对DNA数据存储的“光盘”模式设计编码与数据恢复方法,并结合实测数据开展仿真研究。
在数据存储领域,纠错编码是保证数据可靠性的重要手段。根据香农信息论的信道容量与信道编码的基本理论,纠错码需要与写入/读出的错误特点匹配,才能实现可靠与高效的数据存储[38-39]。目前,数字通信领域的几种重要纠错码已经在体外DNA数据存储中进行了尝试。例如,数字喷泉码用于纠正寡核苷酸分子丢失造成的删除错误[10],里德-所罗门(RS)码纠正碱基删除与随机错误[12-13],低密度奇偶校验(LDPC)码与RS码构成的乘积码纠正删除与随机错误[15]等。而体内大片段DNA存储的编码方法,采用LDPC码与伪随机序列构成的水印码,针对三代纳米孔测序的高错误率,重点考虑难以处理的碱基插入/删节错误[19]。该方法的编码效率较低,为1.19 bit/bp,距离4碱基{A,T,G,C}表示信息的理论极限密度2 bit/bp仍有较大差距。细胞内的数据存储框架,与针对细菌等微生物的基因组从头(de novo)进行测序组装非常类似,需要测序读段从头组装的过程,需要考虑组装后重叠群(contig)的特点,进一步得到完整的数据。因此,为适配二代高通量测序的高精度、组装的重叠群可能存在缺失片段的特点,同时提高碱基承载有效数据的效率,研究便于扩展的信息编码方法,对降低写入成本、开展大片段DNA数据存储具有重要价值。
基于上述考虑,针对细胞内大片段DNA数据存储,为实现信息编码方法适配于测序、读段组装的错误特点,设计了基于多个高码率里德-所罗门(RS)码的符号交织编码方法;提出数据DNA与自主复制序列(autonomously replicating sequence,ARS)交替镶嵌,构建大片段DNA数据存储一般结构的方法。读取端匹配于二代高通量测序特点,设计了基于不同参数(k-mer)组装多个重叠群、根据ARS定位重叠群位置实现数据段合并、使用高码率RS码的纠删纠错算法纠正残留错误的处理流程。研究方法上,为了给从头合成与测序“湿”实验提供灵活的实验前验证与评估,建立了基于计算机的仿真流程,搭建了扩增与测序模型,利用前期的254 kb存储专用人工染色体的真实测序数据[19]进行校准,对编码方案、恢复方法进行了仿真验证。仿真实验证实,在保证端到端可靠写入与读出的前提下,本方法实现的大片段DNA的数据部分逻辑密度为1.973 bit/bp,即使考虑生物功能单元开销,碱基总体逻辑密度仍高达1.947 bit/bp,高于目前基于寡核苷酸池的存储方法(目前报道的最高密度为1.57 bit/nt[10]),非常接近2 bit/bp,充分说明了大片段DNA存储的优势。
1 大片段DNA数据存储的编码方法
大片段DNA数据存储的逻辑结构设计,不同于基于寡核苷酸池(oligo pool)的数据存储,索引与引物(或类似单元,例如酵母人工染色体中的骨架)所占的比例相对较低,在碱基利用率上具有一定优势[11,19]。数据读取阶段,需要先对测序读段进行从头(de novo)组装,类似新物种的基因组从头(de novo)测序。然后,利用纠错码对残留的错误进行纠正,得到完全无错的数据DNA序列,该过程与传统基因组测序不同。因此,设计大片段DNA数据存储的纠错编码方案,需要与测序读段组装后的错误特点相匹配。同时,与生物研究中的基因组组装要求不同,根据数据存储与读取的特点,面向数据存储的读段组装以及后续处理,需要算法有较低复杂度,能在接近实时的情况下实现数据可靠读出,而基因组的从头组装一般对处理时间的要求并不苛刻。
考虑上述特点,提出基于多个RS码交织编码得到数据DNA单元,进一步与ARS序列交替镶嵌,构建体内数据存储人工染色体,形成高效率的大片段DNA数据存储基本结构。针对大片段DNA的二代高通量测序数据,结合现有的读段组装软件实现重叠群快速组装,利用ARS序列定位重叠群、RS码纠错纠删译码,实现数据的快速恢复,其工作流程如图1(a)所示。本文的大片段DNA设计方法包括以下几个要素:高码率的RS码,交替嵌入的ARS序列以及尺度可变的组合方法。实际流程中,将数据写入大片段DNA,也即DNA的合成组装过程,需要借助酵母实现;数据的复制也是借助酵母自身繁殖的过程;核酸提取与建库等是酵母研究中基本操作。进一步,将酵母人工染色体引入大肠杆菌进行富集或直接对酵母进行操作,提取核酸、建库,得到测序数据。前期工作中,我们使用长度为254 886 bp的人工染色体初步证明该方法的可行性,但在更大的长度,实现人工染色体的分离具有难度,也非常具有研究价值,本文不对该问题进行研究。从大片段DNA的合成组装到二代测序输出,依据信息论的研究范式,一般称其为“信道”,本文采用仿真的方法描述该“信道”[图1(a)]。该仿真的“信道”是经过前期254 kb存储专用人工DNA序列的测序数据训练校准的,更接近真实实验,这是本文研究的特色之一。

图1 面向大片段DNA数据存储的高码率RS码编码方法Fig.1 Encoding scheme using a very high code rate RScodes for data storage with large DNA
1.1 多个极高码率的RS码符号交织的编码方法
提出的设计方案中纠错码采用RS码。设计方案与大片段DNA数据存储流程中的错误类型能实现较好匹配。RS码是一种高效、可同时纠正删除(erasure,或称为“擦除”)与随机错误的多进制循环码,可获得理论上最大的最小距离(minimum distance),称为最小距离最大可分(MDS)码;同时,高码率的RS码,冗余符号所占比例较低,编译码复杂度较低,可支撑数据存储的快速译码读出[39-40]。正是由于这些特点,经过优化处理的RS码在硬盘、光盘、固态硬盘以及分布式存储等领域都获得了广泛应用[41-47]。借鉴RS码的成功经验,本文针对二代高通量测序错误率低、能高效组装成重叠群的特点,设计了极高码率(R=0.987)的多个RS码的交织编码方法,并基于此方法构建DNA数据存储单元。
数量不等的DNA数据单元与不同的ARS和载体结合,构造了可变长度的大片段DNA数据存储结构。不同的ARS组成了可选的ARS序列集合[48],根据目前的相关研究结果,ARS的数量较多,能满足本文的设计方案。ARS序列集合在本文的设计中有两个作用:一是与流程中的“湿”操作有关,面向写入侧的实际体内组装与扩增,支持大片段DNA在酵母体内的可靠组装、复制[19,35-37];二是流程中的信息处理,在数据读取时,作为组装的重叠群的标志(类似“路标”),确定组装的重叠群在整个人工染色体大片段DNA中的位置,便于实现数据恢复。
编码流程如图1(b)所示,具体包括以下步骤:
步骤1:数据扰码。也即将数据与已知的伪随机序列叠加[7]。由于数据可能存在长的连续的“0”或者“1”,采用扰码能减少连续比特的数量,从而减少后续长连续碱基的数量,降低测序与合成的难度,减少难以处理的碱基的插入与删节(insertion/deletion)错误[2,10,12]。
步骤2:RS码编码。选择的RS码为定义在有限域GF(212)上的RS码(4095,4040,t=27),其码长为4095个符号,信息位部分的长度为4040个符号,可以纠正55个符号删除或27个符号错误,码率为0.987。每个符号包含12 bit,一个RS码的码字包含的比特数量为49 140 bit。该RS码所定义的有限域为GF(212),阶数较高,但是仅包含55个冗余符号,码率非常高,考虑到RS码的编译码复杂度与冗余符号的数量直接相关,采用该参数的RS码具有可行的编译码实现复杂度,复杂度远低于文献中采用的冗余符号数量高达65 536×15%、定义在GF(216)的RS码[12,39]。
步骤3:多RS码符号交织。根据选择的RS码字的数量P,将其按照列的方式进行排序,然后将其分解为若干组P×P的单元,对每一个单元分别按照图1(b)所示的对角循环的方式进行交织,得到P个数据分组。每个数据分组包含P个RS码符号,从而实现了符号交织,每个分组的大小为4095个符号。在图1(b)中,仅用5个码字的交织为例展示原理[17,39]。在本文的仿真案例中P=100。
步骤4:将RS码码字转化为比特分组。每个数据段对应的4095个符号,转化为49 140 bit,为一个基本分组。
步骤5:比特分组转码为DNA数据序列。按照相邻两个比特转化为1个碱基,来自一个GF(212)上的一个符号转化得到的12 bit,映射为相邻的6个碱基。采用该种转化,有利于RS码发挥其纠正突发删除能力强的优势。一个RS码码字转化为24 570 bp的DNA数据序列。
步骤6:DNA数据序列与ARS、载体等组合,构成完整的大的环状DNA。选择长度较短的P-1个ARS序列,然后按照交替组合的方式,进一步添加载体骨架序列,得到一个环形染色体序列,作为大片段数据存储的基本单元。
1.2 细胞内数据存储的大片段DNA的通用编码设计
在我们以前的工作中,初步验证了酵母人工染色体用于数据存储的可行性与稳定性。在本文提出长片段DNA数据存储的一般框架:选择P个DNA数据段与自主复制序列(ARS)交替镶嵌组合,进一步添加载体,构成一种酵母内数据存储通用大片段DNA结构。该方法灵活选择编码DNA数据单元与ARS的数量,也可在一定范围内改变数据单元大小、数据单元承载有效数据量的大小(也即改变RS码的码率),构成一个规模与效率都可变的长DNA数据存储统一框架。设计中,编码DNA数据单元可能出现ARS序列相似度非常高的情况,但是出现概率较低。选用的100组ARS序列的最小长度为57 bp,则理论上数据DNA部分出现该序列的概率非常低,约为1/457,因此在数据处理中无需对该问题进行处理。其余ARS序列的长度均高于57 bp,出现的概率会更低。
第一个可变参数为人工环形染色体包含的数据单元数量P,在确定每个数据单元的大小后,可以根据数据量以及大片段DNA的合成组装策略灵活选择单元数量。第二个可变参数为RS码的信息符号数量,为进一步提高恢复的可靠性,可以减少每个RS码包含的信息符号的数量,提高RS码的纠错能力。还可以将组装使用的测序数据覆盖度为约束,确定错误率,以此来调整RS码的参数。进一步,本设计结构的各个要素,例如编码方法、ARS单元等均具有可扩展性。例如,也可以采用其他的纠错编码方法构建数据单元,从而匹配采用不同写入或读取模式的需要。利用纠正插入与缺失错误的编码方案,设计了与本文方法类似的结构,用于三代纳米孔测序场景[19];可根据宿主菌的情况,灵活设计复制起始位点(origin of replication,酵母中为ARS)集合、载体序列,从而构建适合不同宿主菌的编码方案。
作为一个例子,本文中我们采用了定义在有限域GF(212)上的RS码(4095,4040,t=27)的构建的编码方法,可以满足设计长度为几十万到几百万碱基的人工染色体(图2)。具体展示了三个设计实例:第一是2 489 847 bp的长序列的方案,存储了一张照片和一份中文文本;第二是两条1.25 Mb的长序列的设计,分别存储了一张照片以及用于填充的文本文件;第三是10条大约250 kb的长序列的设计,该长度与我们之前的实验验证研究相似,相关结论可以借用。根据数据单元的组装结构,可得到该编码方法的编码效率与逻辑密度。本方法采用的RS码码率为R=4040/4095。数据部分逻辑密度为2 bit/bp×4040/4095=1.973 bit/bp。在第一种方案中,考虑镶嵌的ARS序列以及载体序列,总体逻辑密度为1.947 bit/bp。在其他两种方案中,由于载体所占比例增加,总体逻辑密度略有下降,见表1。上述逻辑密度均高于目前文献中四碱基编码的逻辑密度。本文提供的编码方法与数据恢复方法,可在该逻辑密度下实现可靠数据读取,非常接近4个碱基存储数据的理论逻辑密度,也即2 bit/bp。

表1 不同编码方法的碱基逻辑密度比较Tab.1 Base logical density using different encoding schemes

图2 不同数量数据段组合构建不同长度的大片段DNAFig.2 Building of variant-length large DNA integrating different number of data blocks
2 数据恢复策略:ARS导引的重叠群合并与RS码纠错纠删方法
针对提出的大片段DNA数据存储结构,设计了面向二代高通量测序的数据恢复方法。大片段DNA数据存储的读取,与新物种的基因组测序、从头组装非常类似,目标均是得到“完美”的、没有任何碱基错误的基因组。新物种的基因组从头测序,对实时性要求低,可对参数反复调整以得到最优结果[49-55]。大片段DNA数据存储的读出,对算法实时性要求高,传统的生物信息学处理流程并不适用。针对这一特点,数据恢复时,无需在读段组装步骤获得“完美”序列,利用纠错码纠正组装后的残留错误,降低数据读取的整体复杂度,但是需要实现纠错码及其译码方法与组装方法的适配。
本文提出的方法面向数据DNA长度在Kb到Mb级。该长度的DNA适用于常用的二代测序双端读取(paired-end)读段的高效组装,例如基于de Bruijn图的组装方法[53],典型的组装软件有Velvet[54]或ABySS[55]等。组装得到的序列依据内嵌的RS码可实现纠错,得到“完美”人工染色体序列。该方法与传统基因组测序组装的主要差别是:可以在较低的测序覆盖度下得到“完美”的基因组序列,并且在设计大片段DNA时,在数据段避免了重复序列、长连续碱基等,读段组装与恢复方法更为有效。
基于上述思路,提出的数据恢复方法如图3所示,具体步骤为:

图3 基于短读段从头组装、ARS引导的多重叠群合并、RS码纠错纠删的数据恢复流程Fig.3 Data readout processes
步骤1:利用Velvet或ABySS等二代序列组装软件在多个不同长度的k-mer值下实现二代测序读段的组装,得到一组重叠群;该过程同时实现了基于de Bruijn图的数据预纠错,能纠正二代测序中存在的单碱基替换、插入与缺失错误。
步骤2:识别出每个重叠群中的ARS序列,根据ARS序列确定数据读段的位置。ARS位置的识别依据包括插入与删节错误的编辑距离,在本文中我们采用了一种鲁棒的识别策略,只要识别序列与ARS序列的编辑距离小于该ARS序列长度的20%,判断为该ARS存在。识别出ARS位置后,将ARS两侧对应的测序读段,放入该数据段对应的缓存区,直到所有包含ARS序列(或部分ARS序列)的读段被全部标记与分配完毕。
步骤3:对每一个数据读段所对应的部分测序读段,进行大数合并,得到每条数据读段的合并序列。如果某部分读段不存在测序数据,则标记该部分片段为符号删除,如果在某些位置,无法进行大数判决,也标注为删除;进一步将其转化为RS码符号序列。
步骤4:根据分组交织顺序对P个数据段进行解交织,得到P个存在错误与删除的RS码码字。
步骤5:解交织得到RS码的P个码字,分别进行纠错、纠删除译码,得到数据段。
步骤6:根据RS码的译码得到的数据段恢复原始文件,实现比特到文件的恢复。
提出的数据读取方法有以下几个显著特点。首先,使用基于de Bruijn图的不同软件和参数的组装方法得到的重叠群具有一定独立性,对大片段DNA的不同部分有不同的覆盖度。本文中,为降低读取复杂度与读取成本,我们采用低覆盖度的测序数据,例如20×到30×。在低的覆盖度下,不同的k-mer值产生的de Bruijn图的结构有很大的差别,进一步考虑到后续处理方法不同,会得到差别很大的一组重叠群。传统的基因组的组装目标是得到大的重叠群,本文的组装目标是得到尽可能多的重叠群去覆盖数据部分。因此,借用通信中的“分集合并”(diversity and combination)的思想,充分利用重叠群的多样性(diversity),可提高数据段的覆盖度。二代测序读段的错误率本身并不高,因此组装的重叠群的错误率往往较低,RS码需解决的主要问题是缺失部分数据的恢复。
然后,利用镶嵌在数据段之间的ARS序列实现每个重叠群的位置判断,ARS序列充当了一种分布式路标,实现了与大片段DNA数据存储的特点较好匹配。从生物功能方面,该设计也使得大片段DNA在酵母内的组装与传代更为稳定[19,35]。最后,多个重叠群利用ARS定位并合并后,由于ARS缺失或者所有重叠群不能覆盖某区域,会造成数据的某些部分的缺失,将大数判决后存在缺失数据部分标注为删除(erasure),可以充分发挥RS码纠删能力,提高纠错效率。进一步采用交织与解交织,可将组装后数据段中的大段序列缺失转化为随机符号的删除错误,防止单个数据块译码失败[39,47]。这是本文设计的交织的RS码方案的特色所在,较好地匹配了组装后重叠群的特点,实现了整体优化。前已提及,本文未对DNA组装以及随着酵母增殖进行复制的过程进行建模。事实上,DNA组装过程一般采用能保证完整性与正确性的方法。在酵母增殖过程中,在254 kb长度的人工染色体中碱基出错的概率非常低,测试了100代的12个样本中未在数据DNA部分观察到任何碱基错误;但是,长度达到2.5 Mb的人工染色体,超过目前酵母承载外源DNA实验结果的上界,存在不确定性,可能会存在大片段的缺失。针对大片段的丢失,目前的设计方案最大可容忍33 000 bp的大片段缺失。存在大片段缺失时,整个序列会发生整体移位(shift),考虑到ARS序列是分布式嵌入的,基于ARS的重叠群的定位仍可以工作,这也是提出的分布式嵌入ARS序列的优点。
本方法的另一特点是测序与译码的复杂度较低。提出的恢复方法可以在较低的测序覆盖度下完成数据恢复,因此需要缓存处理的总测序数据量较少,从而使得组装处理、重叠群合并等步骤的处理复杂度较低。进一步,采用的RS码码率很高,校验符号的数量仅为55,根据RS码的特点,其译码复杂度较低。设计中通过纠删除与交织技术,充分挖掘RS码的纠错能力,仍能实现在20×测序覆盖度下实现可靠恢复,整体复杂度较低,能在较高的效率实现数据可靠恢复。
仿真实验中,假定人工染色体的测序数据与宿主基因组数据是分离的。该条件可以通过生化操作或测序数据预处理实现。生化处理可根据人工染色体的特性将其分离,在前期针对254 kb的实验中,将人工染色体转入大肠杆菌进行富集。但是,在更大规模的人工染色体,例如Mb长度级别的人工染色体,转入大肠杆菌的方法存在困难。将Mb长度级别的人工染色体分离的操作仍然需要根据人工染色体与宿主染色体之间的关联,并进行进一步设计,这也是目前我们正在开展的工作。在测序数据预处理方面,可开展宿主与人工染色体的混合测序,然后先将测序数据与宿主菌的已知基因组进行比对,再将基因组数据剔除。优点是该方法处理准确度较高。缺点是增加测序数据处理的总量,例如酵母的基因组的碱基数量大约为12 Mb,与设计的2.5 Mb的序列相比,数据量大约是人工染色体序列的4.8倍。
3 实验结果与分析
本文设计了一个长度为2.5 Mb的用于数据存储的酵母人工染色体作为仿真测试实例。高通量测序过程利用二代测序数据的仿真软件ART[56],得到了双端读取的PE150测序仿真数据。然后,开展从测序读段的数据恢复实验,验证提出的大片段DNA编码方法在二代高通量测序下的优越性,也即实现了测序数据特点、从头组装方法以及纠错编码的匹配,从而能凭借非常小的编码冗余实现了非常高的逻辑密度。本部分主要介绍基于仿真测序数据的测试验证方法。
3.1 仿真测序数据校准与分析
本文建立了基于计算机的长DNA片段数据存储仿真平台,如图4所示。目前长片段DNA存储框架中,基因组合成与组装过程产生的错误远少于测序产生的错误。因此,在数据恢复中需要应对的错误主要来自高通量测序。仿真实验中,选择产生测序读段的ART软件模拟测序过程。本研究虽未开展直接的合成与测序实验,我们利用前期的“湿”实验数据[13]对本文的仿真方法进行了校准与验证,使得仿真结果具有较好可信度,一定程度上实现了“湿”实验与仿真设计的融合,使得仿真过程更为合理。进一步,我们分析了仿真的测序数据与端到端的存储恢复性能。

图4 基于计算机仿真的编码大片段DNA体内存储验证流程Fig.4 Verification procedures using computer simulation for proposed encoding method and construction scheme of large DNA chunks in living cells
数据存储的大片段DNA与物种的基因组存在一定差别。为利用ART软件产生更符合实际情况的测序数据,我们先采用以前构建的数据存储人工染色体的二代测序数据[19]对ART软件进行参数训练。具体参数训练与校准中,针对254 886的人工染色体,我们采用的实际测序数据的覆盖度超过200×。然后,利用训练的参数生成针对本文设计的大片段DNA的二代测序数据。我们独立产生了10组30×的数据并开展10次独立实验,总的测序数据量也达到了总覆盖度超过300×。图5和图6给出了生成的仿真测序数据与我们前期实验得到的254 kb人工染色体测序数据的对比。图5比较了读段中测序错误的数量。从该图可以看出,发生错误的读段的数量在20%左右。仿真产生的读段的质量略差于实测数据,这可以更好地说明本文提出方法的纠错能力。图6比较了读段中测序与处在读段中位置的关系。可以看出,二代测序读段中包含插入、缺失与替代错误,错误率在10-4~10-3左右。插入与缺失的错误率明显低于替代错误概率。从图中可看出生成的测序数据特征与实际二代测序数据特征是非常一致的,这说明利用仿真的方法生成测序数据是具有较好可行性的。考虑到目前大片段DNA的组装仍然是非常有挑战性的任务,本文的仿真方法可以在实验前提供更为全面的评估,提高实验效率。
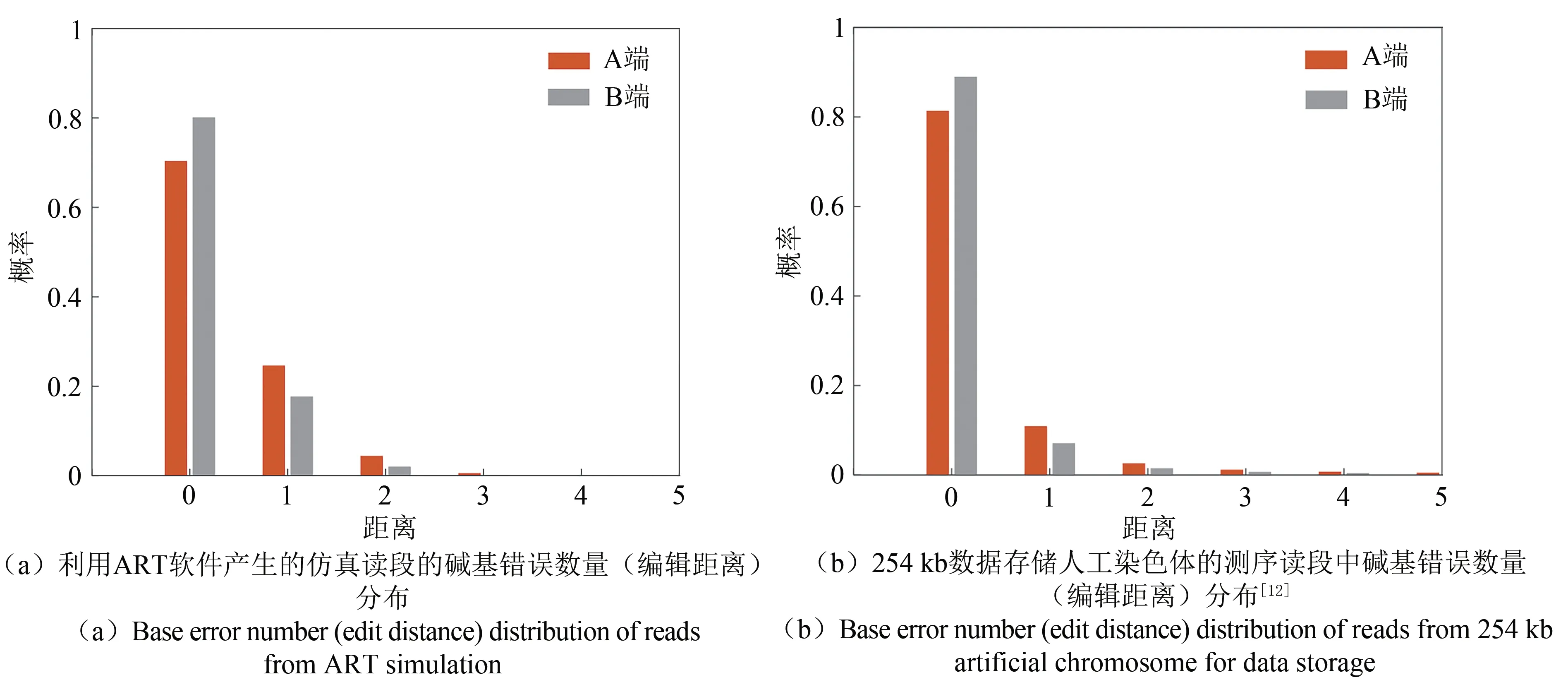
图5 仿真读段与实际测序读段的碱基错误数量(编辑距离)分布Fig.5 Base error number(edit distance)distribution in simulation and real sequencing reads.

图6 仿真与实测读段的碱基错误随着位置变化情况Fig.6 Base error position distribution in simulation and real sequencing.
3.2 译码恢复性能与分析
以100个RS码字的2.5 Mb的基因组为例开展仿真研究。在该模型中,影响数据恢复性能的主要参数是测序数据覆盖度,我们在不同的覆盖度下对编码方案、数据恢复方法进行了仿真测试与分析。测序覆盖度(coverage),体现了对用于存储的DNA的处理复杂度,与成本、信息处理硬件设备复杂度等密切相关。一般而言,二代高通量测序是基于合成的测序方法,测序覆盖度越高,读取成本会越高,测序时间会越长;高覆盖度的测序读段越多,需要的数据缓存的硬件复杂度与计算量都迅速增长。因此,本文参照传统的信息存储设备的特点,致力于在相对较低的测序覆盖度下,实现没有任何碱基错误的快速、“完美”的数据读出。
测试中我们选用覆盖度为20×、25×、30×,每个覆盖度用ART生成10组独立测试数据,进行10次独立的平行组装与译码实验。选用的组装软件为Velvet与ABySS,每种组装方法选择若干不同k-mer值,表2给出了具体仿真结果。图7给出了不同测序覆盖度下的组装错误特性说明,证实提出的多RS码交织并执行纠删纠错方案的合理性。从图7可以看出,当测序覆盖度增加时,多软件多参数的组装方法的性能不断改善,残留的错误与删除数量快速下降,从而可以实现可靠恢复。在测序覆盖度为20×时,单独的Velvet与ABySS组装的错误率均在该方案的纠错能力的边缘(纠正1.34%的符号删除,或者纠正0.66%的符号错误,图7),存在数据恢复失败的情况,详见表2。考虑到组装与重叠群合并策略的波动较大,图7给出了每次实验的具体结果(如图中的“×”和“□”)。
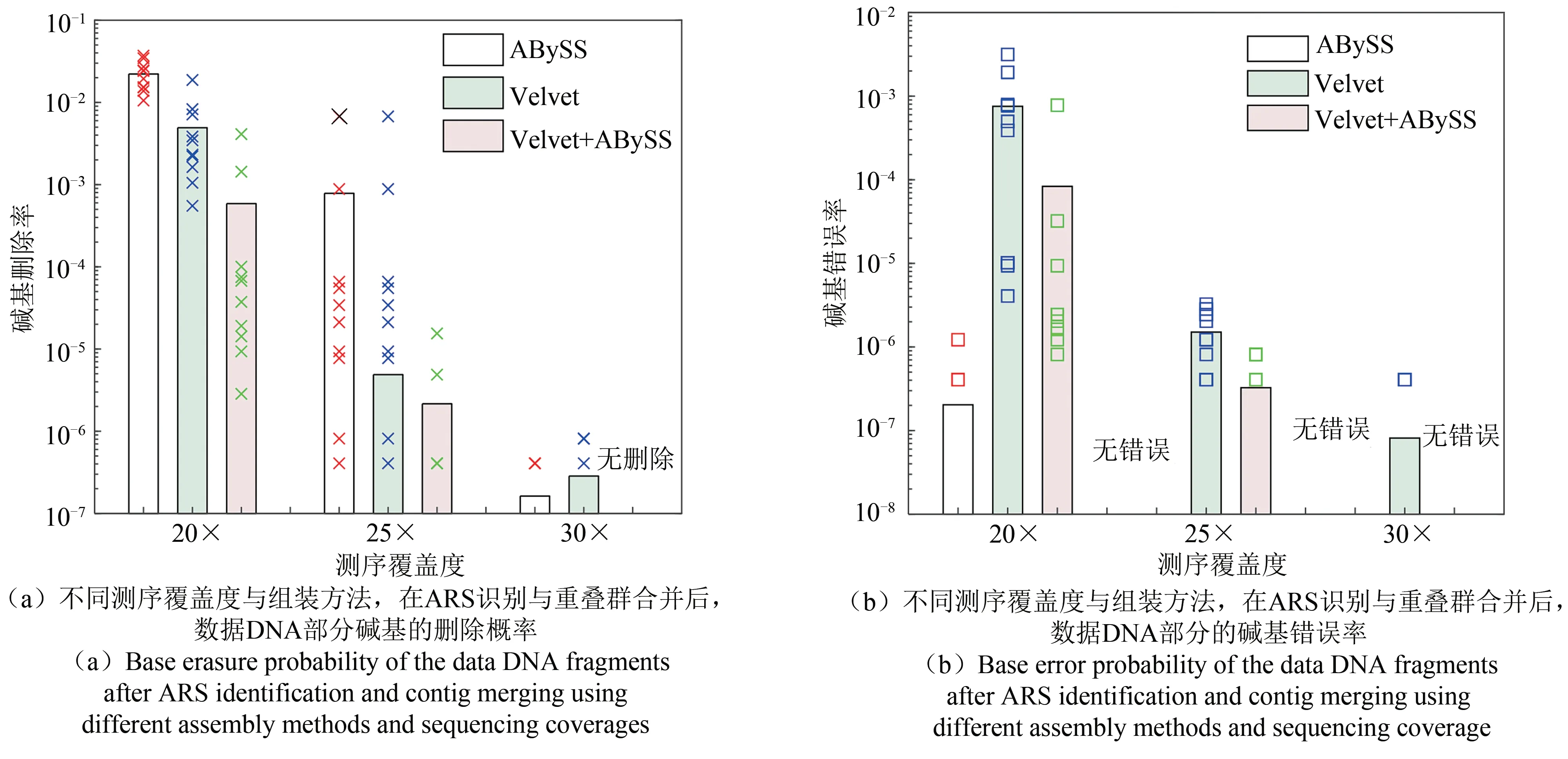
图7 不同测序覆盖度与组装方法,在ARS识别与重叠群合并后,数据DNA部分错误分布Fig.7 Base error distribution of the data DNA after ARSidentification and contig merging using different assembly methods and sequencing coverage

表2 采用交织多个RS码的数据恢复分析Tab.2 Data recovery analysis using interleaved multiple RScodes
从表2的仿真结果可以看出,当覆盖度为25×和30×时,所有方案的10次平行实验均译码成功,验证了该编码方法与数据恢复方法的鲁棒性。同时也可看出,采用多方法、多k-mer与采用单k-mer的结果相比较,读段组装的性能有明显改善,片段缺失与错误均明显减少。表2中仅列出了大片段的错误情况,这些组装、ARS识别后的错误经过交织,可充分利用多个高码率RS码的纠错能力,获得非常高的成功率,实验测试中在25×与30×下数据恢复都是成功的。表2中还列出了若不采用交织方案,仅采用相同参数的RS码独立编码每个数据段不能成功译码的所有情况。若不采用多RS码交织,每个数据块采用单独的RS码编码,由于高码率的RS码的纠错纠删能力有限(纠正Nerasure=55个删除,或者Nerror=27个错误,或者2×Nerror+Nerasure≤55),会存在某些片段不能正确译码的情况,不能完全恢复数据。
在20×测序数据下,采用单个k-mer组装的重叠群错误率很高,不能正确译码,本部分数据量较大,未在表2中列出,详细的信息见本文在期刊官网html文件的补充材料表。但是,多个k-mer值组合的情况仍获得较好的性能。首先,采用Velvet软件多k-mer组装,10次独立实验,仅有第三次会发生解交织后的译码失败,其他情况均正确译码。进一步,在Velvet与ABySS混合组装中,所有10组独立实验,均获得了增益,解交织后全部译码成功。
进一步,根据表2中的数据(第4列)也可看出,本文提出的框架,二代高通量测序读段组装后的错误主要是大片段的数据缺失错误,标记为删除。这是本文主要设计出发点:实现纠错编码、组装方法与测序方法的匹配。第一,考虑到RS码有理论上最优的纠正删除错误能力,本文提出采用RS码来纠正这些突发删除,可取得非常好的效果,能凭借较小的编码冗余度获得可靠数据恢复,可以实现高逻辑密度存储的可靠存储。第二,多RS码交织避免了某些数据段缺失过多无法恢复的情况。
4 结语与展望
为利用细胞内数据处理与存储的优势,本文提出一种针对大片段DNA数据存储的融合码率为0.987的RS码与符号交织的高效编码方法。提出的编码方法实现了将数据文件编码到多个DNA数据单元,DNA数据单元进一步与ARS交替镶嵌组合,构建了灵活的细胞内DNA数据存储结构,实现数据在大片段DNA中的存储。进一步,基于二代高通量测序读段、采用提出的数据恢复方法,可实现可靠的高效率DNA数据读出。该方法实现了交织的多RS码的编码方法、大片段DNA逻辑结构、二代高通量测序、从头组装方法的多要素匹配,从而能够实现非常高的碱基逻辑密度,总体逻辑密度达到1.947 bit/bp,高于目前的主要设计方法,非常接近理论的2 bit/bp。
基于前期的实际生物实验与香农信息论的经典研究方法,提出的大片段DNA数据存储的设计方法,实现了编码方法与细胞内大片段存储信道的匹配,将信息论的研究方法扩展到了合成生物学领域。后续研究将把更多的系统影响因素纳入考虑,例如三代测序、碱基识别方法、测序条形码[57-58]等,为研究者提供更全面、更准确、更系统化的大片段DNA数据存储仿真平台,为研究更接近传统存储系统形态的DNA存储提供依据。
该存储模式中,酵母内大片段DNA的从头合成与组装的“湿”实验是目前技术难度最大、成本最高的部分[27-37]。设计的Mb级别的DNA是否适合在酵母内合成与组装,组装难度如何,都是值得进一步深入分析的问题。之前的实验研究中,仅完成了254 kb的大片段DNA数据存储的全流程实验验证,在一定程度上证明本设计思路具有可行性[19]。到目前,针对已存在的基因序列,可以构造2 Mb以上的酵母人工染色体(YAC)[59-60]。但是,对于来自数字世界转化来的Mb级别以上的DNA数据序列,尚无严格的实验证实。因此,如何进一步突破单个细胞内的存储长度,挑战数据存储的容量上限,并研究其与宿主细胞的相互影响,尤其是Mb级别的完全外源的人工DNA的组装、复制稳定性以及与生物本身基因组的相互作用等问题,都需进一步实验研究。在合成生物学“设计-构建-测试-学习”的闭环策略中,针对数据存储专用的人工染色体,在254 kb正在初步完成该闭环策略[19]。进一步通过“学习”能否构建Mb级别的细胞内存储机制,本文只是完成了“设计”步骤,后续还需要更为深入研究,包括构建、稳定性测试分析等工作。同时,在外部数据体内存储的场景,大片段DNA在细胞内的处理(例如组装、分离等),是合成生物学的重要基础问题[25,32-37],期望在未来取得更大的进展,不仅推动DNA数据存储的发展,也促进合成生物学本身的发展。
针对细胞内长片段DNA存储(“DNA光盘”)的应用场景,考虑到目前长片段DNA的组装成本高,类似早期只读光盘的发展,可通过大量用户共享一次数据写入的成本(“母盘”的成本)才能获得应用优势。考虑到用于存储数据的染色体,借助酵母增殖的复制成本低,能效高,是一种极为高效的生物计算模式。前期研究已初步说明了该类染色体可以有效承载数据,能随着生命过程快速复制,并且便于读出,证明该模式适合数据大规模分发。数据分发(例如通过介质克隆或网络传输),都需要一定成本,利用生命过程的数据大规模复制与分发,即使与基于寡核苷酸池的DNA存储相比,在成本方面仍具有优势,其量化评估需要综合考虑的因素较多,可以将其作为未来研究的方向。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
传染病信息(2021年6期)2021-02-12 01:52:58
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
科学之谜(2019年3期)2019-03-28 10:29:44
发明与创新(2019年9期)2019-03-26 02:22:48
科学之谜(2018年8期)2018-09-29 11:06:46
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
