
Chamaeleo:DNA存储碱基编解码算法的可拓展集成与系统评估平台
2021-07-21 09:30:52平质张颢龄陈世宏倪鸣徐讯朱砂沈玥
合成生物学 2021年3期
平质,张颢龄,陈世宏,倪鸣,徐讯,朱砂,沈玥,5
(1深圳华大生命科学研究院,广东 深圳 518083;2深圳市合成生物学创新研究院,中国科学院深圳先进技术研究院,广东 深圳 518055;3广东省高通量基因组测序与合成编辑应用重点实验室,深圳华大生命科学研究院,广东 深圳518120;4(广东省)华大基因合成基因组学院士工作站, 深圳华大基因科技有限公司,广东 深圳 518120;5深圳国家基因库,广东 深圳 518120;6英国牛津大学大数据研究所,牛津 OX3 7LF;7英国TAICHI AI Ltd., 伦敦 N1 7GU)
作为一种备受关注的新型数据存储介质,DNA分子与传统存储介质相比在信息容量、存储时间及维护投入等方面都极具优势。尤其是随着DNA合成和测序技术的快速发展,基于DNA的数据存储在编码方法与全技术集成系统方面均有了相当的进展[1-2]。如图1(a)所示,常规的DNA存储流程包括从数字文件的编码,生成编码DNA序列的从头合成,通过测序确定DNA文件的序列信息读取信息,根据编码方法进行解码以恢复原始数字文件。其中,作为实现DNA数据存储的关键步骤,编解码方法的开发在过去十年间积累了大量的研究基础。自2012年起,该研究方向聚焦于提升编码密度、通过控制DNA序列的特定生化参数提升DNA存储与现有技术的兼容性、提升编解码方案的稳健性,George Church、Goldman、Grass、Erlich等研究团队提出了不同策略的DNA存储编解码方案[3-7]。在兼容性方面,极端(极高或极低)的GC含量或单碱基长串重复对现有上下游衔接技术都非常不利,会造成DNA合成困难以及DNA测序错误。因此,Church转码采用两种碱基信息对应一种二进制信息的方式,尽管编码密度在所有算法中较低,但其利用随机替换的方式均衡GC含量并避免较长的单碱基重复[3]。Goldman转码利用霍夫曼(Huffman)编码将二进制信息首先转化为三进制信息,再利用轮转编码的方式生成DNA序列以避免单碱基重复出现[4]。Grass转码则利用伽罗瓦域(Galois field,GF)进行编码,使最长单碱基重复长度不超过3[5]。DNA Fountain转码方法首次引入了通信编码中的喷泉码,使得其净信息密度尽可能逼近香农极限,并通过有效性筛选筛除了GC含量不均衡或最长单碱基重复长度超过4的DNA序列,使得生成的DNA序列与现有技术的兼容程度大幅提高[6]。Yin-Yang转码利用2条二进制序列通过不同规则组合成1条DNA序列的方式,通过有效性筛选,使输出序列的GC含量、最长单碱基重复长度以及DNA二级结构自由能都在指定范围内[7]。在编码稳健性方面,为了应对存储过程中可能出现的错误,Grass转码通过引入Reed-solomon(RS)纠错码,增加了编码的稳健性,这种添加纠错码的策略被后续的许多转码方案用来确保数据的准确性[6-9]。在编码学领域也有基于受限编码(constrained coding)相关理论对DNA存储的信道模型进行理论研究的相关报道[10-11]。当然,这些编解码方案也存在局限性。在遇到某些特殊数据结构时,Goldman转码仍然会生成GC含量极端的DNA序列,同时,由于碱基信息间相互关联,单个编码位出现错误,存在后续编码位也有发生连锁错误的风险。对Grass编码来说,GC含量不均衡仍然是其主要风险。DNA Fountain由于其编码方法的特性,在追求极高编码密度的同时,存在有些数字文件无法被编码生成对应DNA序列的问题,或DNA文件无法解码导致完全损坏的风险。因此,该领域研究的研究重点也从以往追求高信息存储密度和技术兼容性的实现,逐渐在编码方法对不同文件的通用性以及高稳健性等方面进行了拓展。
然而,目前已开发的多种转码方法所采用的编程语言、编码所设定的技术参数和标准各不相同,不利于基于已有研究基础的后续开发优化,从应用端看针对不同类型的数据文件最适配的方案选择也缺乏相应的评价与选择标准,从而对促进该领域的交流与发展带来了阻碍。因此,对于已开发的不同转码方法的系统评价标准应当形成明确的共识,并建立相应标准化的体系。此外,现有的转码方法在应对不同数据结构的文件或在不同应用场景下时,表现亦有差异,通过建立系统集成评价平台,针对不同类存储需求以灵活调用的方式提供相应的存储方案也将促进更多DNA存储应用的普及。同时一个开放性、可拓展的集成系统也将有利于该领域的研究学者基于前人研究基础持续提升优化,逐渐形成领域共识,促进这一新兴领域的规范发展。
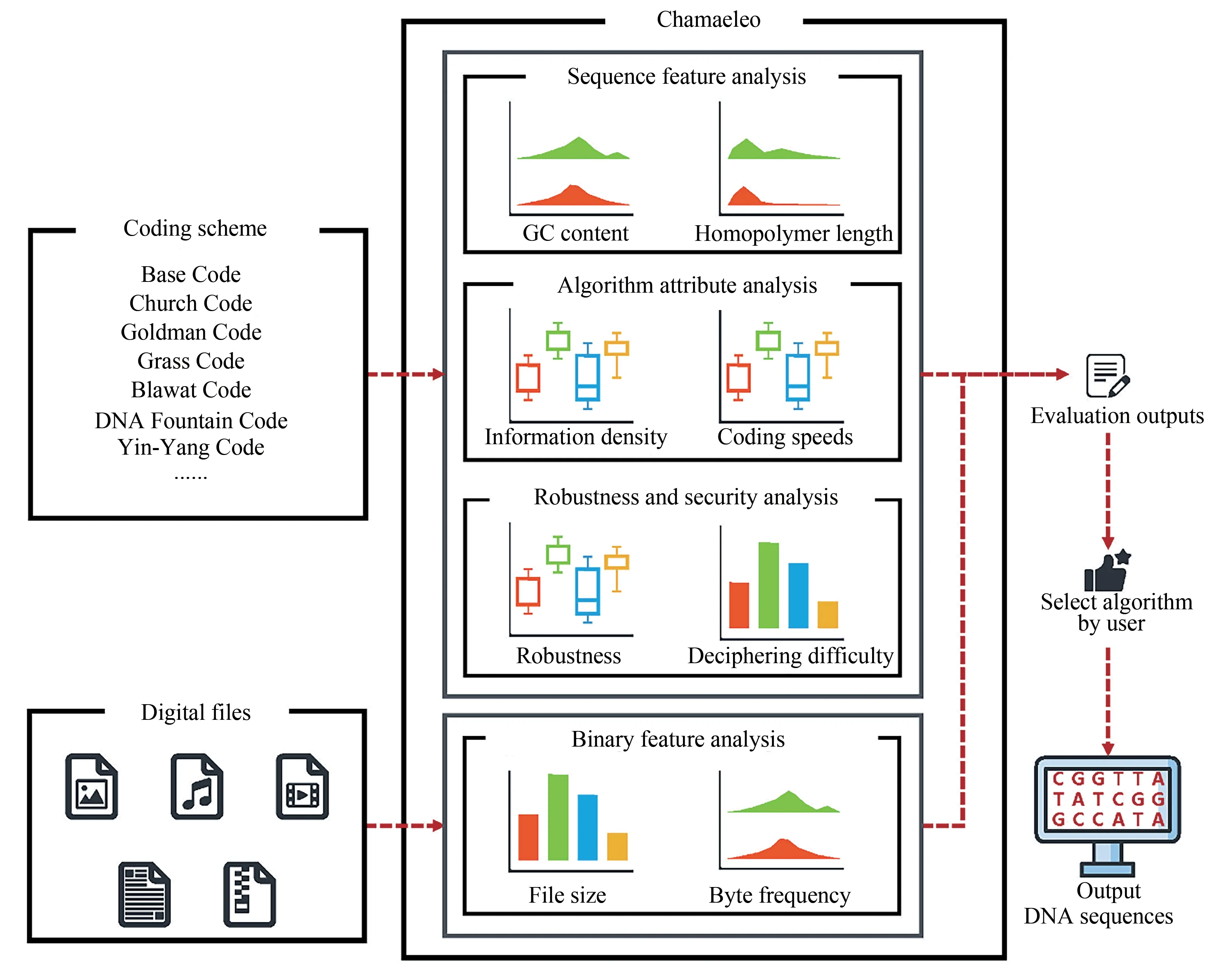
为解决前文所述的问题与需求,本研究开发了一个用于评价与输出推荐编码方案的系统集成软件平台Chamaeleo[图1(a)],针对不同类型的输入文件对模块化集成在该平台中的已报道经典转码方法提供了相应的评价方案[图1(b)]。通过选择此前经典转码方案关注的编码密度、GC含量以及最大单碱基重复长度,对输出序列进行定性定量的分析统计。进一步地,通过建立存储过程中的出错模型和序列丢失模型,对存储过程进行模拟,并据此分析转码方法的稳健性。针对未来某些特殊应用场景,Chamaeleo也提供了不同转码方法所对应数据加密性的理论分析。此外,转码方案的编解码计算效率也将作为额外的参数进行统计,为实际应用提供指导。考虑DNA存储领域还处于快速发展初期,Chamaeleo采用开源、可扩展的设计思路,以支持未来新转码方法的持续嵌入,以及新评估参数的加载。针对特定文件类型,Chamaeleo可通过多维度的分析,对拟采用的转码方法进行较全面的评估,从而为基于特定需求的最优转码方案的选择提供依据。
1 Ch am aeleo平台概述
Chamaeleo作为一个DNA存储转码方法的集成与评估平台,主要功能定位于基于同一文件量化分析不同转码方法之间的特征参数系统性评价并提供方案选择指导。如图1(c)所示,Chamaeleo平台分为三个主要的模块:转码模块、纠错模块和流程模块。
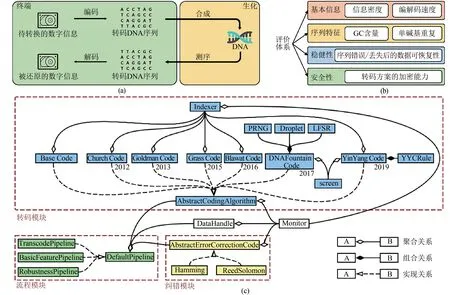
图1 Chamaeleo平台简介[(a)DNA存储的常规流程:该流程分为终端部分和生化部分,Chamaeleo针对DNA存储中的转码步骤进行设计,串联上下游DNA合成与测序;(b)Chamaeleo搭建的评价体系,从多维度对转码方案进行量化分析;(c)Chamaeleo的程序架构及模块间的相互关系。Chamaeleo开源软件平台可通过https:∥github.com/ntpz870817/Chamaeleo进行访问,或通过pypi:pip install Chamaeleo的方式在计算机终端进行安装和使用]Fig.1 Brief introduction of Chamaeleo[(a)General procedure of DNA storage includes in-silico transcoding and experimental DNA synthesis and sequencing.Chamaeleo focuses on in-silico transcoding part and connect the related technologies.(b)The evaluation system created by Chamaeleo quantitatively analyzes coding schemes from multiple aspects.(c)Software architecture and relations between modules and classes.The source code is available at https://github.com/ntpz870817/Chamaeleo,which can bealso installed by thecommand of pip.exe,"pip install chamaeleo"]
转码模块集成了6种经典的转码算法,包括Church转码算法[3]、Goldman转码算法[4]、Grass转码算法[5]、Blawat转码算法[12]、DNA Fountain转码算法[6]和Yin-Yang转码算法[7]。此外,为提供评估指标的基准参考,Chamaeleo提供Base coding作为基准转码算法(即使用A-00、C-01、G-10、T-11的无约束的映射关系进行转码的算法)参与到现有的转码算法的评估体系中。基准算法的评估可以表征无约束的和带有约束的转码算法对特定类型或格式文件中编码得到序列的兼容性差异。
为使转码方法易读并保持后续新开发编码方法可拓展集成,本研究对已开发原始转码算法进行了系统性重构[13]。所有经典转码算法都通过抽象转码算法(abstract coding algorithm)实现Chamaeleo平台系统中的特定接口[14]。未来开发的转码算法也可通过该路径实现对应接口的建立或通过继承平台上已实现转码算法进行特定接口的修改,以完成代码层面的快速搭建(如表S1)。

表S1 使用代码调用流程完成转码过程实例Tab.S1 Example commands to invoke transcoding process
纠错模块采用转码模块相同的架构,目前包含了DNA存储转码方案中最常用的两种纠错码:Hamming码[2]和RS码[5,15]。 通 过 抽 象 纠 错 码(abstract error correction)的接口,Chamaeleo实现了纠错码与校正序列信息两种功能的嵌入。同时,纠错模块在处理比特序列之上,实现了扩展接口,以满足二维及以上比特矩阵的处理。
流程模块则用于进行实际转码/评估任务的执行,转码模块中的转码算法以及纠错模块中的纠错码都会通过实例化的方式被流程模块中的具体流程所使用。通常,一个流程会包含至少一个转码算法以及根据用户来确定是否采用及指定所采用的纠错码。除了最基本转码流程,流程模块还包含三个统计评估流程:算法属性分析流程、序列特征分析流程和转码稳健性分析流程(见下文“评估体系”),用于转码方法的系统性分析,为后续基于不同文件采用的最适配转码方法提供参考依据。
为了进一步增强实用性,Chamaeleo平台亦编写了如数据操作工具(data handler)和流程监控工具(monitor)等DNA存储所常用的工具。数据操作工具支持读/写命令行字符串或文件、压缩/解压二进制信息、保存或加载转码算法/纠错码/流程模块。流程监控工具用于订制化地监控算法或流程执行过程中的完成进展,以及记录特定的过程信息。
2 评估体系
Chamaeleo平台建立了转码方案评估体系,对已集成转码方案进行系统评估,主要涉及算法基本信息、兼容性和稳健性这三方面的系统性评估分析。为建立可比较的评估策略,该平台提供了基准测试文件和基准转码算法,用于收集评估信息并进行统计分析和对比。在基准测试文件方面,考虑到不同的文件具有不同的数据特征,继而会对不同转码方案的实际性能产生影响。因此,该平台目前选择了10个基准文件并尽可能覆盖更多的实际数据特征。基于操作系统最常用的文件类型[16],基准测试文件被分为5类:图片、音频、视频、文本和可执行文件(或压缩包)。由于文件越大,寡核苷酸库中的单条序列所需索引长度就越大,同时包含特殊数据特征的可能性也就越大。而文件比特频率的分布会对DNA存储的转码产生影响,例如,当特殊数据(如大量重复的“0”或“1”)的比例较高时,转码算法不易得到与现有技术兼容性较好的DNA序列。同一类型的文件的不同文件格式会使得信息的比特频率分布出现差异(如图S1图片、音频、视频)。同种格式文件,因为内容有差异,也会导致比特频率的分布出现差异(如图S1文本类)。而如果采用了压缩算法,比特频率的分布将趋向平均[17](如图S1二进制类),但需要承担压缩包损坏导致文件无法恢复的风险。综合以上因素,本研究中所选的不同测试文件在文件格式、文件大小或比特频率上存在显著差异以确保评估体系的可参考性。
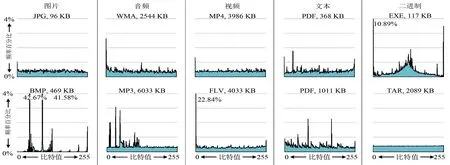
图S1 10个典型测试文件的文件大小和字节频率[每个分布中的4条横线从下到上表示1%~4%的出现比例。部分比特在文件中的出现概率超过4%,最高可达42.67%(BMP代表文件中的比特“00101110”)]Fig.S1 Sizes and byte-frequency distributions of 10 typical test files[The 4 lines in each distribution indicates 1%to 4%from bottom to top.The probabilities of occurrence of few specific bytes exceed 4%,which are labeled with digits.The highest probability of occurrence is 42.67%("00101110"in BMPfile)]
Chamaeleo对转码方案的评估目前分为三个维度:①算法基本信息,通过算法属性分析流程统计信息密度、转码所需时间以及转码成功率等;②技术兼容性,通过序列特征分析流程评估转码方案与上下游技术的兼容性,通过收集被转码方案编码(一个或多个文件)获得的DNA序列,统计并分析针对这些DNA序列与上下游衔接技术的兼容性相关参数,比如GC含量和单碱基重复等;③稳健性,由于针对编码DNA序列的合成与测序实际操作过程中,不可避免地会因为所采用的技术本身的局限性而产生错误(碱基插入/替换/删除)或序列丢失。通过转码稳健性分析流程,在转码获得的DNA序列文库中分别随机引入错误和序列丢失,计算源文件的成功恢复率,进而表征该转码方案对错误和序列丢失的可容错性。
2.1 编码密度评估
为降低DNA存储技术的成本,转码方法通常需要在保证数字信息完备的情况下尽可能减少DNA序列合成量,因此,编码密度是转码方法最重要的评价指标之一。在DNA存储中,除了携带源文件二进制信息的数据区,为了确定混合DNA分子中不同DNA序列对应源文件中信息的位置,在编码DNA序列中会引入索引区。DNA扩增是目前DNA存储流程中实现获取部分数据所常用的技术方法,因此也需要设计引物区,结合特定引物对文库进行扩增,以便数据备份和测序建库。此外,为了使这些DNA序列在解码过程中可以一定程度上纠正碱基突变带来的问题,编码DNA序列往往还会选择引入纠错区以提升数据可恢复的稳定性[图2(a)]。基于特定编码设定下的二进制-碱基间映射关系,可以计算出不同的转码方案对应的理论编码密度。但如上所述,除数据区外,DNA序列还会包括索引区、引物区和纠错区,因此实际的编码密度会低于理论编码密度。例如,当信息区的长度固定,伴随着数字信息(文件)的总比特数的增加,其索引区占整条序列的比例会增加,从而导致编码密度的降低。与其他转码方案不同,DNA Fountain转码采用Luby Transform(LT)码[6,18],使用长度为16个碱基的索引区保存每条DNA序列所包含的随机数种子。因此,在数字信息大小不超过500 MB的情况下[6],其实际信息密度不变。而当数字信息的大小大于这个阈值,DNA Fountain转码需要使用更长的索引区(超过16个碱基)去保存每条DNA序列包含的随机数种子。
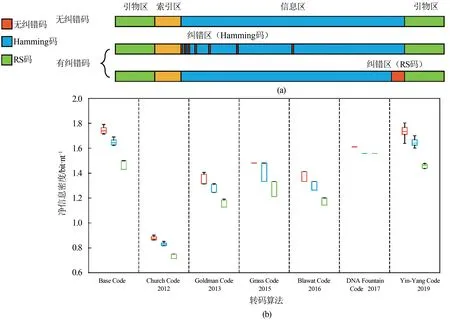
图2 DNA序列结构设计与已收录编码方法的净信息密度评估[(a)输出DNA序列设计,纠错码为可选项,其中,Hamming和RS码在结构上的分布略有不同;(b)测试文件通过不同转码算法获得的净信息密度,其中Goldman转码算法使用的报道中生成的哈夫曼树[4],DNA Fountain转码算法使用伪随机数生成器中默认度分布参数(δ=0.05,c_dist=0.1)以及默认的7%的冗余[6],Yin-Yang转码算法则选择相关研究中采用的第888号规则[7]]Fig.2 Structure of output DNA sequences and their net information densities[(a)Basic design of output DNA sequences with/without optional error-correction codes(Hamming code and RS code).(b)Distribution of net information density using different coding schemes.The setting of parameters is identical to that of original report:For Goldman's coding scheme,the Huffman tree used in the evaluation process is set as the same in Goldman,et al[4].For DNA Fountain scheme,the degree distribution tuning parameter(δ=0.05 and c_dist=0.1)and redundancy(7%)used in the evaluation process is set as the same in Erlich,et al[6].For Yin-Yang coding scheme,rule No.888 wasused as reported in Ping,et al[7]]
此前有相关研究围绕信息密度提出了一些相关计算方法与相应的概念用于评价转码方案的性能[3,6,19]。 如 碱 基 编 码 率 (base coding density,bit/nt)描述的是在不考虑分子拷贝数的情况下,实际操作中一个碱基携带的有效比特数量,即:

而物理信息密度(physical information density,petabyte/gram,即PB/g)则是基于流程操作考量下提出的概念[20],其描述的是经过换算后,实验中每克碱基携带的有效信息量(字节数),即:

此外,还有一些概念,如编码潜力(coding potential)和实际容量(realized capacity,净信息密度与信道的香农容量之比)[6],也相继被提出,但由于它们涉及信息论相关的复杂理论推导,与实际应用层面数据差别较大,因此在DNA存储领域未被广泛认可。参考该领域中如上概念的普遍认可程度,参考引物区长度在不同存储的应用需求下会出现差异的情况,并结合考虑目前一般采用长度为200 nt的寡核苷酸文库作为DNA存储的主要方式(数据区长度约120~140 nt),Chamaeleo平台中选择采用数据区编码256 bit数据条件下的净信息密度(net information density,即输入信息的比特数除以转码完成后排除引物区序列的碱基数)[6]作为评估转码方法编码密度的参数:

在编码密度评估中,本研究基于无纠错码、含汉明码和含RS码这三种情况分别进行了评估。为和此前的报道统一,本研究在二进制信息层面进行纠错码的插入。程序默认原始比特序列长度为128 bit,采用经典设置的(7,4)Hamming码,其校验位长度为8 bit,且校验位插入在原始比特数据的2的幂次位上,理论可以纠正至多1个碱基替换。目前,此前提出的转码算法皆采用的是RS码,通常设置3个字节以纠正至多3个碱基替换错误[2,21]。此外,按照文献的设计,RS码通常会被放到原始比特序列的后面。通过针对目前已开发的6种编码方法进行对应编码密度的量化,我们发现由于测试文件的大小不同,受到索引区长度变化的影响,绝大多数编码方法的编码密度都在一定范围内发生了波动,而DNA Fountain转码由于索引区长度固定,编码密度最为稳定。另一方面,Yin-Yang转码表现出相对较高的编码密度,接近于无拘束的基准转码方法。此外,总体趋势上不同的编码方法在引入纠错功能后均显示出较其理论编码密度有5.19%~13.49%的下降。采用RS码比Hamming码的下降更为显著,说明转码方法的纠错能力越强,越需要支出更多的信息密度[22]。
2.2 兼容性评估
DNA存储中的兼容性体现在与现有技术(DNA合成技术[23]、DNA测序技术[24]、PCR扩增技术[25]等)的适配程度。极端(极高或极低)的GC含量或单碱基长串重复对现有上下游衔接技术都非常不利,会造成DNA合成困难以及DNA测序错误,进而导致数据无法恢复的问题。因此避免极端GC含量和单碱基长串重复是目前各转码方法重点关注的方向。其他可供选择的评价参数还包括DNA序列的二级结构自由能、规律性重复序列与特殊序列(如酶切位点、毒性序列)的出现频率等。
在兼容性评估实验的过程中,由于添加纠错码在一定程度上能够消除部分对转码算法兼容性不友好的数据特征[26],本研究中以不采用纠错码的方案作为兼容性下限的评估。
如图3所示,针对测试文件所涉及到的数据特征,对比基准算法,6个转码算法都对兼容性做了相应的优化。依据目前的测试结果,已收录的转码算法都回避了单条序列可能存在的极端GC含量。其中,DNA Fountain转码算法和Yin-Yang转码算法将GC含量控制在了40%~60%,在兼容性方面表现较为突出。而Church转码算法、Goldman转码算法、Grass转码算法以及Blawat转码算法转码所获DNA序列都存在40%以下或者60%以上GC含量的情况,因此存在生成DNA序列与上下游技术不兼容的风险。Yin-Yang转码算法的平均GC含量分布在40%和60%附近相比DNA Fountain转码算法更低,因此相比DNA Fountain转码算法,它更容易进一步提高GC含量的限制。控制单碱基长串重复方面,无约束的基准算法所获序列的单碱基重复长度存在于1~42之间,而已收录转码算法对其都有严格的限制。由于Goldman转码方法的单碱基重复设置为1,因此其单碱基重复的峰值聚集在1的区间,而其他方法的单碱基重复并没有较大的差异,都处于2~4之间,说明目前所有的转码方案都在控制单碱基长串重复方面具备良好的兼容性。
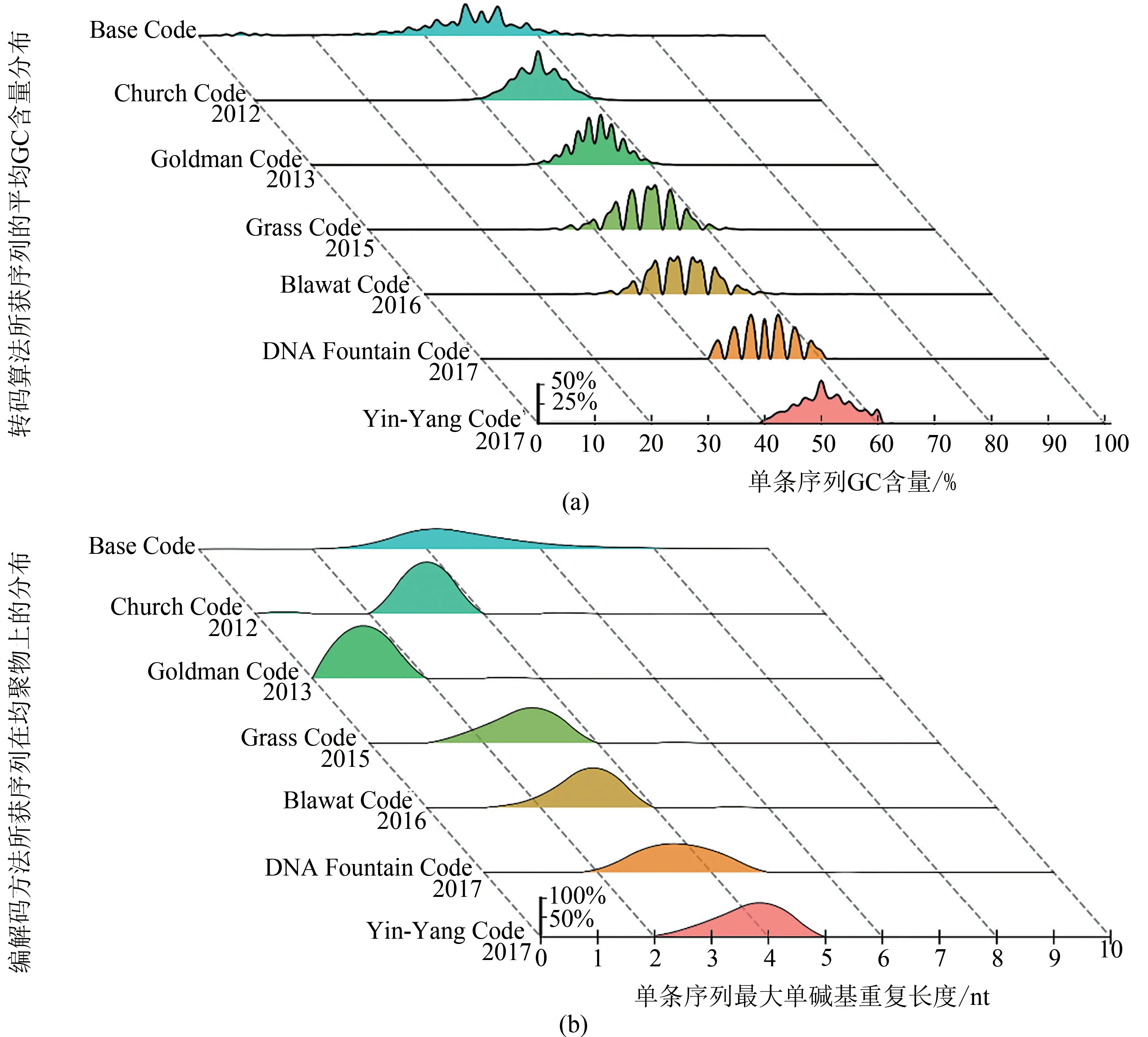
图3 转码方案的兼容性评估[在无纠错码情况下,不同转码算法从10个测试文件中编码所得DNA序列的GC含量分布(a)和最大单碱基重复长度分布的统计(b)]Fig.3 Compatibility evaluation of coding schemes[Distribution of GCcontent(a)and maximum homopolymer length(b)of DNA sequencesfrom transcoding of 10 test-filesby different coding schemes.For Basecoding in(b),themaximum homopolymer length is42 and not shown in thispanel for aclarity purpose]
2.3 稳健性评估
由于成本与访问速度的限制,DNA存储目前仍然被认为适用于长期的冷数据(无需频繁访问的数据)存储,同时它可以应用基于宿主菌的体内存储或寡核苷酸库的形式进行体外存储。而在这些存储过程中由于宿主菌自身可能发生的变异积累又或者合成过程引入的错误,DNA分子不可避免地会发生碱基插入、删除、替换等变异以及自然降解。因此,如何从转码方案层面应对这些可能带来信息丢失的风险尤为重要。在转码后得到的DNA序列文库中随机引入定量的碱基错误和序列丢失,再使用对应方案进行解码,Chamaeleo通过收集和计算所得正确解码信息对原始信息的覆盖率作为稳健性评估的指标。
如图4(a)所示,当引入1%的序列丢失后,大多数转码方案的数据恢复率都在98.98%~99%之间。DNA Fountain转码与其他转码算法相比,单个文件的数据恢复率(恢复出的正确数据量/源数据的总量)波动较大(11.75%~99.99%)。其原因在于:为了满足筛选条件,DNA Fountain转码算法所获的每条DNA序列(编码数据包)通常会包含多条比特序列信息,同时这些数据包之间具有相互关联的拓扑结构,当一条DNA序列丢失后,可能会造成更多DNA序列无法满足解码条件,从而丢失更多的比特序列信息。Yin-Yang转码算法的数据恢复率在98.95%~99%之间,其恢复率下限略低于除DNA Fountain外的其他算法。在Yin-Yang转码算法中,一条DNA序列包含2条比特序列信息,所以当单条DNA序列出现错误不可修正,其损失可能会是其他转码算法(Church转码算法、Goldman转码算法、Grass转码算法、Blawat转码算法)损失的2倍。对任一转码方案,利用大量物理冗余或逻辑冗余均可以很好地应对引入的错误。以DNA Fountain转码算法为代表的抹除码,依据其编码原理特征,只要在解码阶段接收到足够的编码数据包,源数据即能完全恢复。值得注意的是,由于喷泉码中的度分布设置,单个编码数据包与其他源数据分片存在相互关联,因此当逻辑冗余不足的情况下,解码过程中单个编码包的错误或丢失可能会连锁影响其他编码包的解码,而多个编码数据包的丢失可能造成整个文件几乎无法恢复。另外,直接对源数据进行编码的转码算法,例如Church转码算法,在添加源数据量10%的逻辑冗余后,转码算法对于<10%的序列错误或丢失将具备较强的耐受性,然而不能保证源数据的完全恢复。
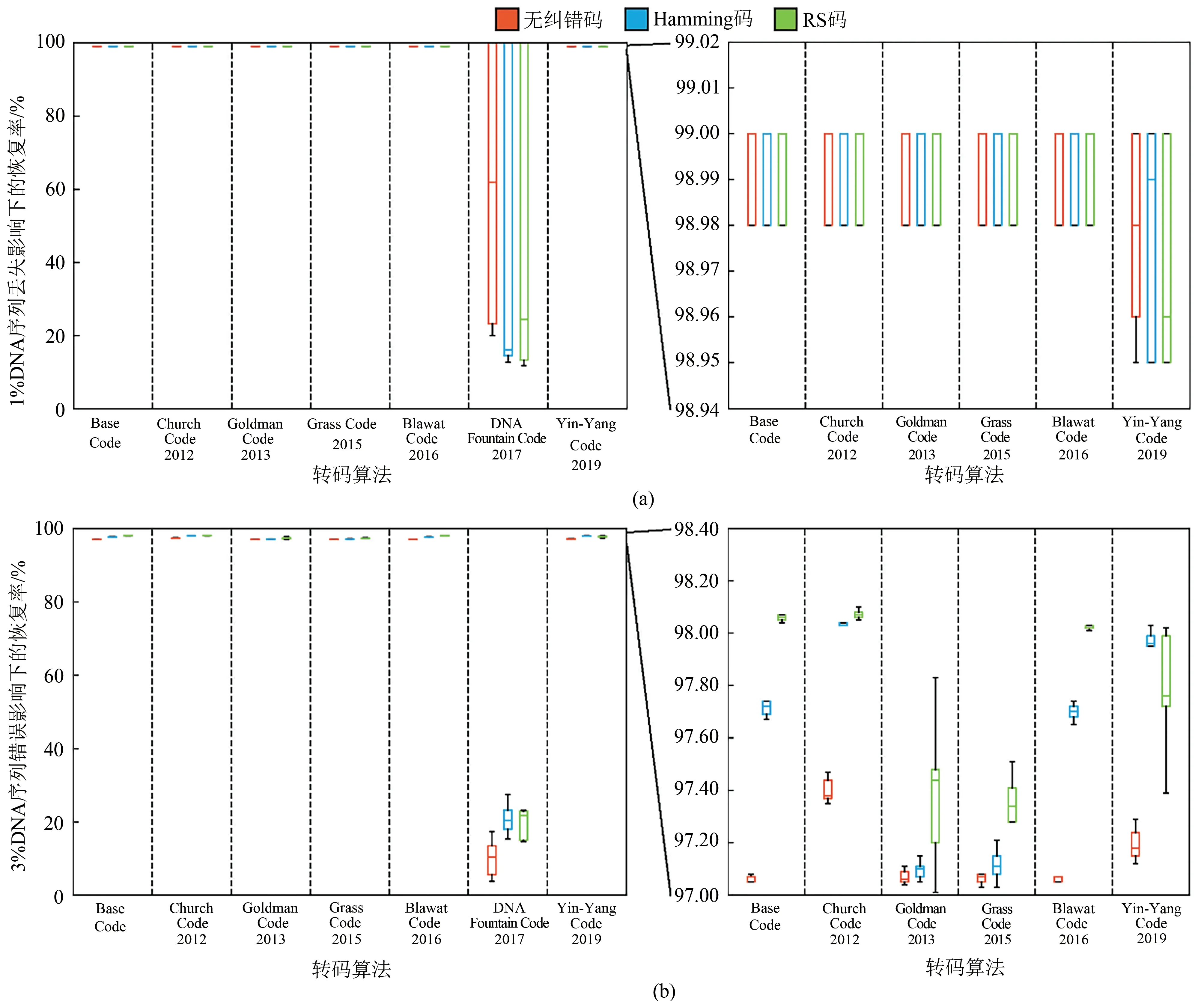
图4 转码方案的稳健性评估[(a)在测试文件转码获得的DNA序列文库中引入1%的随机序列丢失,对比解码后文件与源文件,获得该情况下文件的恢复率。右侧图为98.94%~99.02%区间放大。(b)在测试文件转码获得的DNA序列文库中引入3%的随机碱基错误(插入、删除、替换各1%),对比解码后文件与源文件,获得该情况下文件的恢复率。右侧图为97%~98.4%区间放大]Fig.4 Robustness evaluation of coding schemes[Distribution of file retrieval rate under condition of(a)1%random sequence loss and(b)3%of nucleotide error(1%for insertion/deletion/substitution,respectively).Figureson theright isthezoom in part for acloser view]
在DNA存储的生化反应过程中,稳健性评估
需要考虑的因素为由于测序深度、PCR随机性等生化操作造成的DNA分子的突变或者丢失。这些突变和丢失通常分为系统误差和随机误差。在DNA存储中,随机误差一般由测序产生,而测序过程的随机错误通常可以用序列比对的方式进行相互校正,系统误差一般由合成或分子生物学操作产生,无法通过常规测序数据处理方式进行校正。因此,在Chamaeleo评估体系中,这里的稳健性评估指的是系统误差对转码算法造成的影响。根据此前的文献报道[6,20,25],经过测序后,序列的丢失率一般在1%~2%左右,因此本文用1%序列丢失进行稳健性评估。通过常规DNA合成的错误率分析,一般认为错误率为0.3%左右,而大片段DNA组装合成中错误率会更高,因此本文用各1%碱基错误(插入、删除、替换)进行稳健性评估。当引入碱基插入、删除、替换各1%的错误后,大多数转码方案的数据恢复率都在97.05%~98.62%之间。DNA Fountain转码算法在此情况下的文件恢复率较低(3.78%~27.52%),显示其在应对碱基错误的稳健性不足。另外,纠错码的使用对稳健性的提升有较为明显的作用。对所有转码方案而言,纠错码的纠错能力越强,稳健性越好。值得注意的是,目前常规的纠错码,如本文使用的Hamming码、RS码等,仅对碱基替换有效,而由碱基插入或删除导致的错误,常规的纠错码无法进行错误的发现和纠正。但在当前DNA合成流程中,每种DNA序列的合成拷贝数大约为107个,所有拷贝同时出现插入或删除错误的可能性极低,因此,可以通过测序后的序列比对找出共有序列,从而纠正碱基插入或删除错误。
2.4 转码方案的加密应用可能性评估
针对现有编码方法基于特定规则的转码过程,如果规则相对简单易推导,则数据可加密性会较低,相应的有特殊存储需求的加密数据的安全可控性也会较低。对于部分转码算法来说,其比特与碱基之间的映射关系并不是唯一的,如果解码过程和编码过程使用的映射关系不同,就无法获得正确的原始比特信息。因此,这些映射关系可以被视为密码学的秘钥,在基本的转码任务以外,用于特殊信息的加密和解密。不同的转码算法包含的映射关系数量是不同的。通常,映射关系(秘钥)的数量和信息被破译的时间成正相关[27]。本研究统计了已集成转码算法的映射关系数量,用于评估转码方案的数据安全性。
Church转码算法和Blawat转码算法的映射关系是固定的且只有1种。Goldman转码算法的映射关系可以视作Huffman三叉树[28](k+1=3)与字节(n=256)之间的映射关系,其映射关系的数量可以使用Huffman三叉树的形态种类表示。通过弗斯-卡特兰(Fuss-Catalan)数[29-30]进行换算,可得映射关系共有种。Grass转码算法的映射规则是在3个碱基(由于规定后两个碱基不可相同,4×4×3=48个组合)中筛选47个组合作为伽罗瓦域,并将其余比特信息进行映射,所以会存在种映射关系。在DNA Fountain转码算法中,4种碱基可以通过不同的排列方式和2个比特组合(00,01,10,11)进行映射,因此该转码算法包含=24种映射关系。Yin-Yang转码算法由虚拟碱基、Yang规则和Yin规则共同构成。其中,虚拟碱基有=4个选择;Yang规则是在4种碱基中选择2种映射为0,剩下2种映射为1,共计有=6种组合;Yin规则是在Yang规则的基础上进行0和1的分配,共有种组合,因此共有有4×6×256=6144种映射关系。
综上所述,Chamaeleo平台将通过计算转码方案的映射关系数量(秘钥数量),作为其数据安全性的参考依据。
2.5 基于图论的转码算法理论评估
DNA存储转码方案的理论层面研究仍存在空白,结合DNA的生物特性以及合成与测序的技术进行的理论研究也相对较少。因此,除了量化分析实际转码后的参数,本研究尝试从理论层面对转码算法进行评估分析,通过转码算法的映射关系可视化(图5),并使用图论[31]进行理论层面的分析与评估。除了使用信息论[24]宏观描述转码算法的信息密度外,图论分析有助于更细微地剖析转码算法的特征与倾向性。
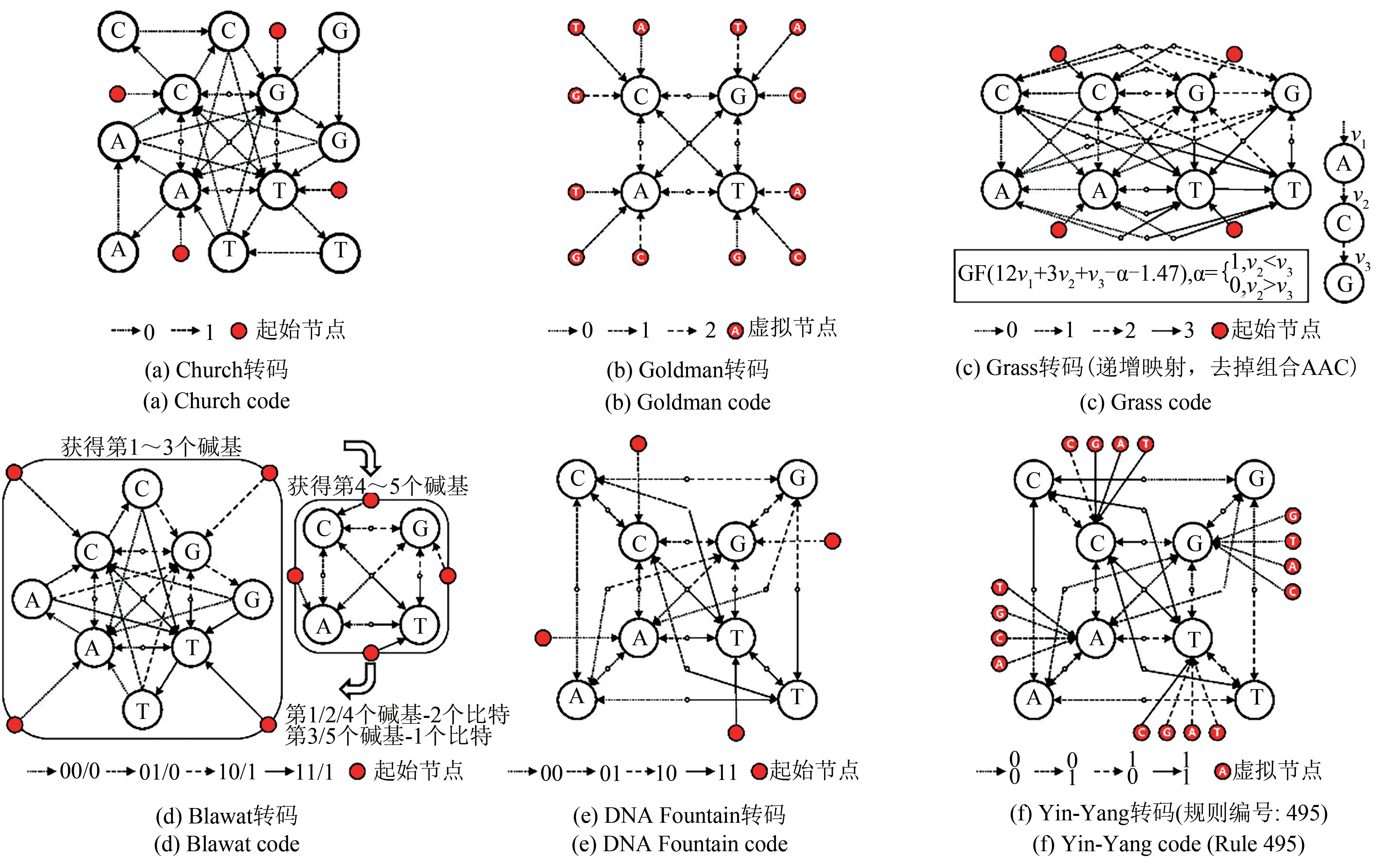
图5 使用图论对不同转码算法进行可视化[实心红点表示起点或虚拟起点(即Goldman转码及Yin-Yang转码中为编码第一位碱基所用的虚拟位)。在每一轮编码过程中,已知当前的碱基(节点)和输入的二进制(箭头),跳转至下一个节点并输出其所指代的碱基,最后将下一个节点作为当前节点。在对应的每一轮解码过程中,已知当前节点(碱基)和下一节点(碱基),获得两个节点之间的箭头,并输出箭头指代的二进制信息,最后将下一节点作为当前节点]Fig.5 Visualization of different coding schemes using Graph-theory-based presentation[Red dot represents starting point or virtual starting point(for Goldman&Yin-Yang coding schemes).During each step of encoding,with known previous base(node)and input bit value(arrow),the current basewould be obtained from graph.During each step of decoding,with known previous and current nodes(base),the bit value(arrow)would beobtained from graph]
对于一个长度为n比特的序列来说,0和1的组合方式是有限的,即2n种。通过转码过程,可以获得转码方案可生成的所有DNA序列。进一步将这些DNA序列作为图的节点,并使用k-mer组装[32]的方式进行连接作为图的边,获得转码算法的图表示。根据生成图的各种属性可以从理论层面分析转码算法的性能,而通过图的出度情况[31],可以近似估计转码算法的净信息密度。图中每个节点的兼容性即为DNA序列的局部兼容性,基于贪心算法[33],局部特征可以反映全局特征,即全长DNA序列的兼容性。通过图中的环结构[31],不仅可以统计单碱基重复的情况,还可以统计重复序列、回文重复等复杂情况。对于图中具有周期性且具有统计意义的子图或模式[34],可以通过计算,例如标准分数(z-score),分析算法对不同比特数据信息的倾向性。这些属性可以结合生物特性进行进一步的分析。
目前基于图论的转码算法映射关系可视化有助于理解转码过程,转码算法可生成的图种类数量亦能在一定程度上反映出转码规则的复杂程度,从而与其在加密存储的应用中数据安全性的性能形成对应关系。
3 结语
DNA存储集编码学、密码学、信息学、分子生物学、生物信息学、计算机科学等多学科高度交叉发展而来,其全流程的实现仍高度依赖DNA合成与测序技术的支撑,相关的理论基础的研究还处于早期阶段,是一个充满想象力的新兴研究领域。其中,DNA存储中的编解码方法是作为连接数字信息和DNA分子的关键步骤,也是过去十年该领域的主要研究方向。不同的编码方法在存储信息密度、技术兼容性与存储稳健性方面各有千秋,但根据不同的存储需求,目前该领域还缺乏方法间标准化比较评估体系的建立,不利于研究人员基于已有研究的再次开发以及DNA存储应用端的普及。
因此,本研究开发了一个DNA存储碱基编解码算法的可拓展集成与系统评估平台Chamaeleo,取变色龙可针对不同环境快速适应进行特征变换之义,旨在促进该领域的开发者进行协同开发,为应用端提供一个辅助的指导工具以实现不同存储需求的应用。Chamaeleo集成了系列已开发的DNA存储编解码方法并对其算法在软件工程层面进行了优化,提供了一个通用的DNA存储编解码应用工具。同时,聚焦DNA存储与现有技术的兼容性以及长期存储情况下的稳定性,通过选取领域内已有共识的关键参数,建立了一套DNA存储编解码方案的评估分析体系。利用不同类型、不同格式的文件对经典DNA存储编解码方案进行多维度评估,得到的数据可以进行后续的系统性分析。值得指出的是,本研究中采用的评估参数是相互关联、相互影响的。例如,编码密度会受到兼容性、稳健性、安全性等方面的影响。从编码学中constrained coding的角度看,拘束条件必然会导致编码密度的降低[11]。为提高DNA存储整个流程的稳健性或保证数据安全性,转码方案一般会采用增加纠错码、多拷贝[4]或者对比特序列预先进行异或操作[35]的策略。但这些策略会导致冗余的增加,一定程度上也会降低转码方案的编码密度。兼容性则意味着转码方法可以通过设置相关参数以降低与上下游衔接技术不兼容序列出现的可能性,以提升转码方案的整体可行性。因此,在选择最优转码方案时,需要对所有指标进行综合考量。我们希望Chamaeleo平台的建立能促进领域内学者的交流以及新研究者的融入,有助于形成标准化的行业流程与评价指标,从而推动该领域规范有序的快速发展。同时,本研究中首次提出基于图论的理论评估方法及“特征”“倾向性”等评价指标,旨在促进DNA存储整体评价体系的发展。在未来,我们期待更多DNA存储领域的研究者将其独特的DNA存储转码方法嵌入Chamaeleo开源工具平台中,也希望能通过广泛的交流与讨论形成更多的有指导意义的评价指标和策略,帮助该领域编解码方法的理论体系逐渐形成。
尽管DNA存储目前已经有了诸多的文献报道,数据存储体量也逐渐逼近GB级别,但相比于传统的光盘、硬盘存储,还需要在很多方面进行突破,Chamaeleo平台也需要相应的完善和优化。从理论层面,结合DNA合成与测序技术特点,通过数学、编码学等对DNA存储的最优转码方法进行研究。转码应用方面,在某些编解码过程中,为解决序列内部的错误和序列丢失问题,设计纠错机制时会设置不同的内码和外码[5],因此在已有纠错模块的基础上,Chamaeleo也将提供内外码设置的选项以提高纠错机制的灵活性。评估体系方面,根据不同应用场景,可以对目前已有评估参数或者新评估参数进行标准化,并设置相应的权重,从而根据实际需求提供最优转码方案。从安全体系层面,需要进一步完善数据安全体系,针对个人隐私安全、数据安全、军事应用等不同场景,建立相应的加密方式,保障数据安全。应用层面,建立高效低成本且功能化的全流程DNA存储体系需要软件平台进一步集成上下游衔接技术参数来进一步提升兼容性方案的输出,同时也需要考虑数据存储架构、快速寻址访问、低成本复写策略等方面的需求。在DNA存储之外,目前也有基于质谱技术,利用寡肽或代谢物进行信息存储的策略被陆续提出[36-37],Chamaeleo平台也可以提供相应转码方案的程序端加载接口,为硅基存储与碳基存储建立桥梁。
综上,基于DNA介质的新型数据存储作为一种具有划时代意义的存储方式,或将打开全球海量数据存储的新纪元。DNA存储技术的发展,既需要针对编解码方法持续取得创新突破,建立颠覆性DNA存储信息学理论基础,也依赖于如DNA合成组装与测序的上下游衔接技术的快速发展以形成高效低成本规模化存储能力。我们希望Chamaeleo平台的建立能作为向该目标发展的一个起点,助力生物技术与大数据信息技术的协同发展,在历史文化传承信息的永久保存、经济大数据的长期保存、军事部署等战略信息的传送和保存等领域发挥作用。
致谢:在程序设计和文章撰写过程中,庄乾龙协助了部分程序模块的填写,哈佛大学Geor ge Chur ch教授、周广宇博士和首都师范大学葛根年教授及兰昭君参与了算法方面的讨论,维也纳工业大学的Nat al ie Fr eiber ger协助我们在IOS平台进行了系统测试,在此一并致谢。
补充材料
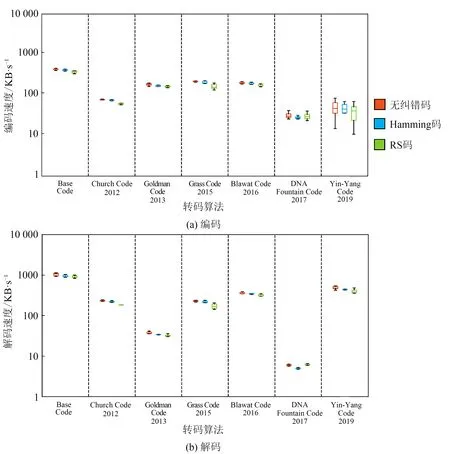
图S2 不同转码算法编码和解码速度的比较(测试环境:Windows 7系统;i7 CPU处理器;12GB内存;Python版本,3.7.3;IDE,PyCham 2019.1)Fig.S2 Encoding and decoding speed of different transcoding algorithms(Test environment:Windows 7 environment including an i7 CPU and 12 GB of RAM using Python 3.7.3 in the IDEPyCharm 2019.1)
猜你喜欢
广东通信技术(2023年11期)2023-12-10 12:28:38
缔客世界(2020年1期)2020-12-12 18:18:28
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
商情(2019年3期)2019-03-29 12:04:52
发明与创新(2019年9期)2019-03-26 02:22:48
财讯(2018年22期)2018-05-14 08:55:57
传播力研究(2018年7期)2018-05-10 09:42:47
现代商贸工业(2016年35期)2016-04-09 06:59:32
