
ACAD9基因新发变异致线粒体复合物I缺乏症20型1例报告并文献复习
2021-07-16 12:03崔清洋曹银利唐成和王卫卫贾倩芳
临床儿科杂志 2021年7期
崔清洋 曹银利 唐成和 韩 炜 王卫卫 贾倩芳
新乡医学院第一附属医院儿科(河南卫辉 453100)
线粒体疾病是一组遗传和临床异质性疾病,一般有进行性和多系统性受累特征,是人类最常见的遗传性疾病之一,发病率估计为1/5000活产儿。线粒体受核基因组与线粒体基因组的双重遗传调控,其功能是通过氧化磷酸化给正常细胞的功能提供所需的能量。而线粒体内的氧化磷酸化系统包括线粒体呼吸链复合物(复合物Ⅰ~Ⅳ)和腺苷三磷酸酶(复合物Ⅴ),复合物Ⅰ缺陷是线粒体病最常见的生化缺陷,占所有缺陷的1/3[1],其中线粒体基因编码13个亚基,而核基因编码约70余个亚基,涉及线粒体DNA的复制、转录、翻译以及酶复合体的组装、物质代谢及线粒体融合相关的1 000余种的编码产物。
人类的线粒体疾病约40%在新生儿期发病,其中25%为母系遗传,大部分为常染色体隐性遗传,时有散发、X 连锁和常染色体显性遗传。有报道核基因变异所致的线粒体疾病占到75%~95%[2],而核基因的变异可分为10 种类型[3],其一类型即为呼吸链复合物亚基及组装因子的变异,而涉及复合物Ⅰ变异的基因包括NDUFS 1-8、NDUFV 1-2、NDUFA 1-2、NDUFA 9-12、NDUFB 3、NDUFAF 1-6、FOXRED 1、NUBPL及ACAD9。
线粒体复合物Ⅰ缺乏症常见的临床表型有Leigh综合征、Leigh样综合征、Leber遗传性视神经病、线粒体脑肌病、乳酸酸中毒和卒中样发作、新生儿心肌病、严重新生儿乳酸酸中毒、白质脑病、非特异性脑病等,其中的Leigh综合征和Leigh样综合征最为常见,占到单纯性线粒体复合物Ⅰ缺乏症的50%以上。
目前累及核基因ACAD9变异的线粒体复合物Ⅰ缺乏症20型(mitochondrial complexⅠdeficiency type 20,MC 1 DN 20)国内外报道很少,仅为个案或小的队列研究,且部分资料不详,目前国内尚无新生儿病例报道。本文回顾分析1例MC1DN20新生儿的临床资料,其ACAD 9基因的c.1278+1 G>A剪切变异和c.895A>T错义变异均为国际上首次报道。
1 临床资料
患儿,男,胎龄35+3周,剖宫产后8分钟,于2020年8 月24 日入院。患儿系G 4 P 4,因母亲羊水过少、易栓症剖宫产出生,出生时无产伤及窒息史。父母体健,非近亲结婚,姐姐9 岁,体健。入院体格检查:一般情况及反应差,肢体活动少,哭声低弱,全身皮肤发绀,皮肤散在胎脂,口周稍发绀,呼吸节律不规则,吸气三凹征阳性,双肺呼吸音偏低,心腹未见异常,四肢肌张力稍弱,原始反射未引出。入院时患儿全身皮肤发灰,予复苏囊加压辅助通气及吸痰肤色转红润。入院实验室检查:血常规白细胞6.2×109/L,红细胞4.97×1012/L,血红蛋白187 g/L,血小板297×109/L;血气分析pH 值 7.26,氧分压91 mmHg,二氧化碳分压28.5 mmHg,钾7.31 mmol/L,钠128 mmol/L,氯89 mmol/L,钙0.55 mmol/L,镁0.97 mmol/L,磷1.93mmol/L,乳酸 12.5 mmol/L,碳酸12.5 mmol/L,剩余碱-13.1 mmol/L。胸片疑似新生儿呼吸窘迫综合征,心影外型增大。血气分析pH值波动于6.85~7.06,乳酸波动于22.0~26.9 mmo/L,碳酸波动于3.5~ 5.9 mmo/L,剩余碱波动于-24.6~-36.3 mmo/L;血乳酸12.5 mmol/L、血氨218.8 μmol/L;超敏C反应蛋白0.33 mg/L,IL-6 14.99 pg/mL;总蛋白48.9 g/L,白蛋白32.6 g/L,总胆红素53.7 μmol/L,直接胆红素12.5 μmol/L,丙氨酸氨基转移酶5 U/L,天冬氨酸氨基转移酶42 U/L,γ-谷氨酰转肽酶366 U/L;乳酸脱氢酶536 U/L,α-羟丁酸脱氢酶371 U/L,肌酸激酶439 U/L,肌酸激酶同工酶25.73 ng/mL;肾功能未见异常;B型钠尿肽前体36 000 pg/mL。血串联质谱及尿气相色谱-质谱示多种氨基酸增高,尿多种氨基酸增高,需要鉴别是否为线粒体疾病,建议基因检测以便鉴别诊断。头颅彩超示第三脑室增宽(约8 mm),余未见异常。入院第3天复查头颅彩超示第三脑室增宽(约10 mm),余未见异常。肝胆胰脾、双肾及输尿管彩超未见异常。心脏彩超肺动脉高压(50 mmHg);入院第4天复查心脏彩超肺动脉高压(76 mmHg),较前升高。
患儿入院后予经鼻持续气道正压通气辅助通气、暖箱保暖、静脉营养、预防出血、抗感染、西地兰强心、多巴胺维持血压、碳酸氢钠纠正酸中毒治疗后代谢性酸中毒难以纠正;入院第5天予包括辅酶Q10、核黄素、维生素E及C在内的鸡尾酒治疗后复查血气分析好转,辅助通气下经皮脉氧饱和度不稳定,予气管插管有创呼吸机辅助通气下经皮脉氧饱和度维持在80%左右;但患儿心率仍反复下降,肝脏较入院时增大,复查凝血功能明显低下,予冷沉淀对症治疗。因患儿病情持续危重,家属放弃治疗后患儿死亡。
因患儿反复高乳酸血症、血串联质谱及尿气相色谱-质谱提示线粒体疾病可能,经医学伦理审核,并获得家长知情同意后,采集患儿外周血 4 mL 及其父母外周血2 mL,乙二胺四乙酸(EDTA)抗凝,送北京康旭医学检验有限公司进行基因检测。
提取患儿及其父母基因组DNA,使用 Illumina 高通量测序仪和 Agilgent 公司的 SureSelect 探针富集体系进行二代基因检测。使用软件 CASAVA(1.8.2)将原始数据转化为可识别碱基序列,然后进行Align 分析、SNP 分析和DIP 分析,获得靶向区域变异位点信息。通过PolyPhen-2、SIFT、Mutation Taster进行蛋白质损伤分析,获得需要进一步验证变异位点。在人类基因组数据库GenBank中获得IDS基因变异位点基因序列,在引物设计网站 Primer Z(http://genepipe.ncgm.sinica.edu.tw/primerz/ primerz4.do)设计并合成引物。对变异位点进行PCR扩增后进行一代测序验证,排除二代测序假阳性位点。
测序分析发现患儿ACAD 9基因第12 内含子c.1278+1G>A杂合剪切变异,该变异导致该基因的编码序列第1278位碱基后的第1个碱基G(位于内含子区域)变异为A,距离该外显子边界(c.1287)1 bp 的位置;ACAD 9基因第9 号外显子上的c.895 A>T(p.Ile 299 Phe)杂合错义变异,导致ACAD 9基因所编码的第299 个氨基酸由异亮氨酸变为苯丙氨酸。蛋白功能损伤预测中c.1278+1 G>A:SIFT:“-”,Polyphen 2“-”,MutationTaster 有可能致病;c.895A>T:SIFT示有可能致病,Polyphen2有可能致病,MutationTaster示有可能致病。上述变异可能导致蛋白质功能受到影响,两个变异均尚未见文献报道(参考数据库:HGMD Pro、PubMed及ClinVar)。该变异不属于多态性变化,在人群中发生频率极低(参考数据库 1000Genomes及dbSNP)。家系验证结果显示,患儿c.1278+1 G>A剪切变异遗传至母亲,母亲为杂合变异,父亲未发现该基因位点变异;c.895 A>T 错义变异遗传至父亲,母亲未发现该基因位点变异;两种变异形成复合杂合变异,符合线粒体复合物Ⅰ缺乏症核基因变异的常染色体隐性遗传方式(图1、2)。
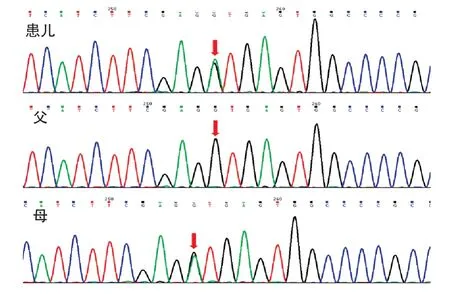
图1 ACAD9 基因c.1278+1G>A 变异测序图
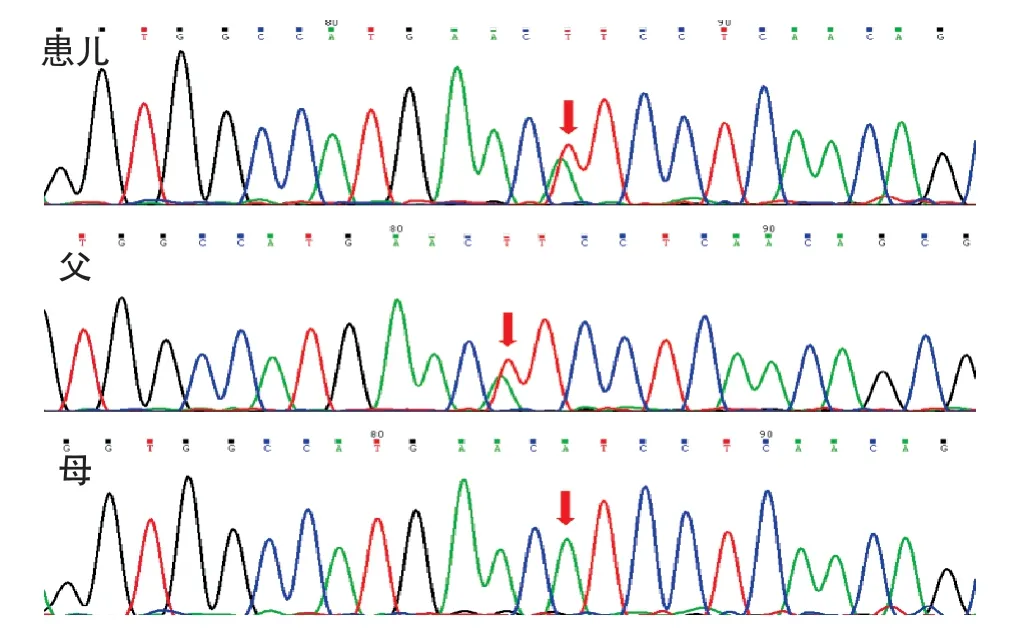
图2 ACAD9 基因c.895A>T 变异测序图
根据美国医学遗传学与基因组学学会联合美国分子病理学会2015 年制订的《基因序列变异的解释标准和指南》进行致病性分析[4]。ACAD 9基因c.1278+1 G>A 的致病性:①c.1278+1 G>A 变异为±1 或 2 位置的剪切变异,为致病变异(非常强致病性证据,PVS 1);②c.1278+1 G>A 变异通过比照ESP 数据库、千人基因组数据库、EXAC 数据库中正常对照人群中未发现(或隐性遗传方式中频率极低)的变异(中等致病性证据,PM 2);③综合上述c.1278+1 G>A变异的证据强度为“PVS1+PM2”,判断为导致受检者发病的疑似致病性变异。ACAD9基因 c.895A>T的致病性:①c.895A>T位于突变热点和/或位于已知无良性变异的关键功能区域(中等致病证据,PM 1);②c.895 A>T 变异通过比照ESP 数据库、千人基因组数据库、EXAC 数据库中正常对照人群中未发现(或隐性遗传方式中频率极低)的变异(中等致病性证据,PM2);③c.895A>T变异经多种算法预测会对基因或基因产物功能造成有害影响的变异(支持致病证据,PP3);④综合上述c.895A>T变异的证据强度为“PM1+PM2+PP3”,判断为导致受检者发病的临床意义不明性变异。但ACAD 9基因c.895 A>T 变异位点SIFT、Polyphen2及MutationTaster均预测为有可能致病。尽管目前尚无文献报道且判断为临床意义不明,但亦未有无致病性的定论,需待日后进一步行相关验证。
结合患儿临床表现及基因检测分析诊断ACAD 9基因c.1278+1G>A和c.895A>T复合杂合变异所致的MC1DN20基本明确。
2 讨论
ACAD9基因定位于3q21.3,基因组全长约33.50 kb,包含18个外显子。目前MC1DN20基因变异类型多以复合杂合及纯合变异为多见。ACAD 9基因编码的酰基辅酶A 脱氢酶9(ACAD 9)是线粒体呼吸链复合体I(complex I,CI)的组装因子,ACAD9基因变异是CI 缺乏的常见原因,ACAD 9 在体外对长链脂肪酸也保持酶ACAD活性,但这一功能的生物学相关性仍存在争议,部分原因是ACAD9的组织特异性表达,在肝脏和神经元中高表达,而在皮肤成纤维细胞中表达最少。研究显示,在表达高水平ACAD9细胞中,酶促ACAD 活性是完全脂肪酸氧化能力所必需,而功能丧失决定ACAD9缺乏患者的表型中很重要[5]。ACAD9缺陷患者的治疗应以治疗CI 与脂肪酸氧化功能障碍为主。
国内曾有报道通过二代测序技术发现1 例因ACAD 9基因所致的线粒体疾病,但患者年龄、性别及具体变异类型不详;另有报道1 例ACAD 9基因c.797G>A(p.Arg266Gln)和c.1359-1G>A复合杂合变异所致的儿童线粒体疾病相关癫痫[6]。
德国曾报道4 例线粒体复合物Ⅰ缺乏症患儿,其中包括2 例同胞[7]。在2 例同胞中,姐姐出生后不久就出现心肺功能下降、肥厚性心肌病、脑病和严重乳酸酸中毒,并在出生第46 天死亡,其肌肉中的复合物Ⅰ活性降低至9%~14%,肝脏中的复合物Ⅰ活性降低至1%,成纤维细胞中的复合物Ⅰ活性降低至32%~39%;弟弟出生时表现低血压、心肌肥大和乳酸性酸中毒,给予积极的核黄素治疗后临床反应良好,5岁时认知障碍,精神运动发育无异常;这2 例同胞是ACAD9基因p.Phe44Ile和p.Arg266Gln复合杂合变异。另2例女童,均有肥厚性心肌病、脑病和乳酸酸中毒,分别在2岁和12岁时死亡,基因检测发现分别为ACAD9基因的p.Ala326Pro和p.Arg532Trp复合杂合变异、p.Arg266Gln和p.Arg417Cys的复合杂合变异。另一家系中3 例线粒体复合物Ⅰ缺乏症患儿,均患有肥厚性心肌病、低血压、乳酸性酸中毒和运动不耐症,其中1例肌活检复合物Ⅰ活性为正常3%,基因测序发现ACAD9基因c.1594C>T(p.Arg532Trp)纯合 变异[8]。
美国曾报道3 例线粒体复合物Ⅰ缺乏症,表现为偶发性肝功能不全,同时伴有慢性神经功能障碍。其中1 例14 岁患儿死于轻度病毒性疾病和服用阿司匹林引起的Reye样发作和小脑卒中,尸检结果显示弥漫性肝微泡性脂肪变性、脑部弥漫性水肿;ACAD9基因检测发现第1个ATG上游44个bp的4 bp插入。1例10岁女孩,4月龄首次出现暴发性肝衰竭,随后出现急性肝功能不全和低血糖。还有1例4.5岁女童,死于心肌病,其同胞在22月龄时也死于心肌病。但这2例未行基因检查。
还有报道来自3个无血缘家庭的9例线粒体复合物Ⅰ缺乏症患儿,包括7例女孩和2例男孩。大多数患儿在新生儿期出现乳酸性酸中毒,并在婴儿期死亡。除先前报道的肥厚性心肌病外,5例患者(来自2个家庭)有动脉导管未闭。家系Ⅱ的2例患儿在儿童时期出现运动不耐受,伴有轻度左心室肥大,在核黄素治疗下临床症状稳定。家系Ⅰ的二代测序发现ACAD 9基因c.1636G>C(p.Val546Leu)纯合变异,家系Ⅱ的二代测序发现ACAD9基因c.509C>T(p.Ala170Val)和c.1687C>G(p.His563Asp)复合杂合变异,家系Ⅲ的二代测序发现ACAD9基因c.1240C>A(p.Arg414Ser)and c.1650_1672dup(p.Leu558Profs*45)复合杂合变异[9]。
上述文献复习证实线粒体复合物Ⅰ缺乏症常导致多系统损害,临床表现多样。本例患儿临床表型主要是顽固性代谢性酸中毒及乳酸性酸中毒,基因检测发现ACAD9基因的c.1278+1G>A和c.895A>T复合杂合变异,支持文献报道,但患儿病情危重,未行头颅MRI检查,且患儿肝脏逐渐增大考虑也与心力衰竭有关。本例患儿虽予碳酸氢钠纠正酸中毒但无明显疗效,而予核黄素治疗后血气分析好转。
尽管尚无法明确ACAD9基因c.1278+1G>A和c.895A>T变异位点的致病性,但患儿有MC1DN20典型的临床表现,且c.1278+1G>A和c.895A>T变异(尽管目前c.895A>T尚无文献报道且判断为临床意义不明,但亦未有无致病性的定论,需待日后进一步行相关验证)不属于多态性变化,在人群中发生的频率极低(所参考数据库 1000 Genomes及dbSNP)。故综合考虑患儿基因检测结果及临床表型,可基本确认ACAD9基因c.1278+1G>A和c.895A>T变异位点为患儿的致病性变异。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
湖南饲料(2021年4期)2021-10-13
现代畜牧科技(2021年4期)2021-07-21
西华大学学报(自然科学版)(2021年3期)2021-05-17
种子(2021年3期)2021-04-12
西华大学学报(自然科学版)(2020年6期)2020-10-15
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
药学研究(2015年11期)2015-12-19
