
进行性家族性肝内胆汁淤积症2例临床及遗传学分析
2021-07-16 12:03何佳倩孙长宇乔芳芳郑利民
临床儿科杂志 2021年7期
何佳倩 孙长宇 乔芳芳 郑利民
1.郑州大学第一附属医院感染性疾病科(河南郑州 450000);2.中山大学生命科学学院 (广东广州 510060)
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)是一组罕见的常染色体隐性遗传疾病,与胆汁酸分泌或运输缺陷相关,通常在婴儿期或儿童期出现渐进性黄疸、严重的瘙痒症,最终进行性胆汁淤积会导致门脉高压、肝硬化、肝衰竭、肝细胞癌[1]。自1969 年首次描述PFIC 以来[2],过去50 年在对PFIC 相关疾病的理解和诊断方面取得巨大进步。目前已经确定6种基因导致不同类型的PFIC:ATP 8 B 1、ABCB 11、ABCB 4、TJP 2、NR 1 H 4、MYO 5 B[3]。肝脏组织病理学检查和基因检测可协助诊断。本研究回顾分析2 例PFIC 患儿的临床特点及遗传学分析结果,以提高临床医师对本病的认识,有助于早期识别 PFIC。
1 临床资料
例1,女,15岁,因皮肤黏膜及巩膜黄染7月余,加重2月余就诊,伴皮肤瘙痒,腹泻白色黏稠样便,4次/d。患儿之前反复出现皮肤瘙痒,无黄疸。患儿系G1P1,足月顺产,出生体质量3 kg,生长发育同正常同龄儿。父母身体健康,无类似疾病家族史。体格检查:身高158 cm,体质量44 kg,全身皮肤黏膜及巩膜黄染,未见肝掌、蜘蛛痣。腹部平坦,肝脾肋下未触及。实验室检查:血常规红细胞3.21×1012/L,血红蛋白(Hb)113g/L,肝肾功能、凝血功能、二便常规无异常,HBsAg、HCV 抗体、HIV 抗体、梅毒螺旋体抗体阴性。腹部彩超示肝脏大小正常,肝实质回声稍致密,脾大(长径133 mm,厚径38 mm),副脾。家属拒绝行肝脏穿刺病理学检查。
例2,男,3 岁6 月龄,因皮肤黏膜及巩膜黄染1 月余就诊,无发热、腹泻等症。患儿为双胞胎,系G1P1,足月剖宫产,出生体质量2.6 kg。生长发育同正常同龄儿,出生后较其胞弟易哭闹,常出现头部摩擦枕头现象,自其可以语言表达(约1 岁6 月龄左右)后间断诉全身瘙痒,无皮肤黏膜及巩膜黄染。每3~6个月复查肝功能,瘙痒发作时丙氨酸氨基转移酶(ALT)、天冬氨酸氨基转移酶(AST)、胆红素稍升高,瘙痒间期基本正常。患儿父母、双胎弟弟身体健康,无类似疾病家族史。体格检查:身高95 cm,体质量 17 kg,全身皮肤黏膜及巩膜黄染,未见肝掌、蜘蛛痣。腹部平坦,肝脾肋下未触及。实验室检查:AST 50 U/L,总胆红素173.6 μmol/L,直接胆红素139 μmol/L,间接胆红素34.6 μmol,总胆汁酸396 μmol/L;铜蓝蛋白47.45 mg/dL,血常规、肾功能、凝血功能无异常,HBsAg、HCV 抗体、HIV 抗体、梅毒螺旋体抗体、风疹病毒抗体、单纯疱疹病毒抗体、CMV、EBV、自身免疫性肝病抗体均阴性。磁共振胰胆管造影示肝脏大小、形态正常,肝内胆管显示可,肝内胆管未见明显扩张。肝脏穿刺病理切片内见9个中小汇管区,部分间质少数单个核细胞浸润;肝小叶结构清晰,肝板较完整;多数毛细胆管扩张,内见较大胆栓,中央静脉周围尤著,少数肝窦内见脱落的胆栓(图1)。
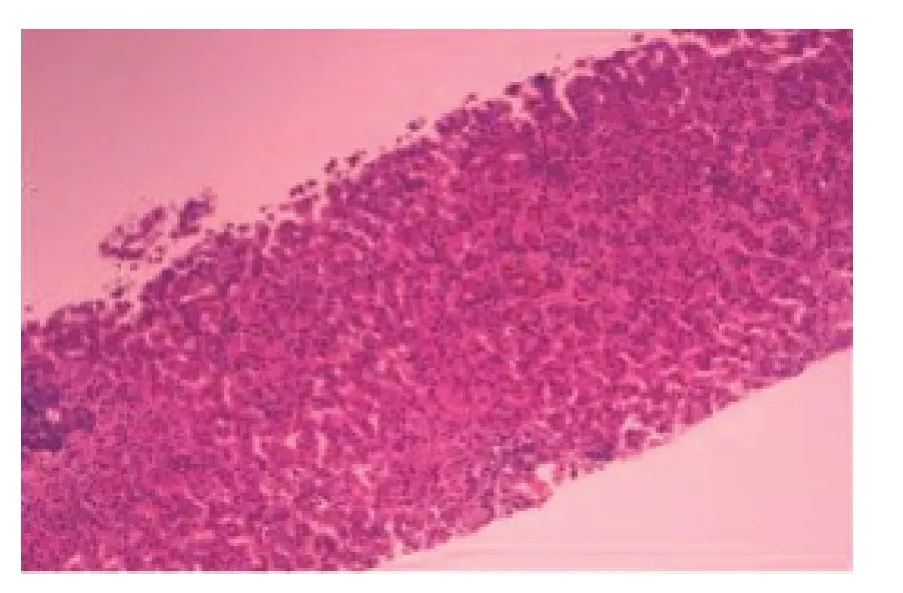
图1 例2 患儿肝脏组织病理(HE×400)
因2 例患儿临床表现考虑PFIC,为进一步明确诊断,经医院伦理委员会批准、家属知情同意对患儿进行全外显子基因检测。采用EDTA 抗凝试管,抽取患儿及其父母外周血各2 mL。天根血液基因组DNA提取试剂盒(TIANamp Blood DNA Kit)提取基因组DNA,制备样本DNA 模版。文库构建后使用PE 150模式在Illumina(San Diego,CA)测序平台上进行双端测序,并根据基因检测结果进行Sanger测序分析。结果显示,例1 患儿ATP 8 B 1基因存在复合杂合变异,c.2652G>C和c.1573C>T,其中c.2652G>C遗传自父亲,导致第884 位密码子由编码赖氨酸变为编码天冬酰胺(p.Lys 884 Asn);c.1573 C>T 遗传自母亲,导致第525 位编码精氨酸的密码子变为终止密码子(p.Arg525Ter)(图2)。c.2652G>C变异位于保守的HAD-like domain功能域,参与蛋白之间相互作用,生物信息学软件SIFT 和Polyphen 2 预测其分别为有害的(Damaging)和很可能有害的(Probably Damaging)变异;检索数据库及文献发现其未见报道。c.1573C>T为无义变异,可能会导致编码的蛋白功能丧失,有文献报道其曾在胆汁淤积患者中检出。一代验证结果表明,患儿携带的变异c.2652 G>C 与c.1573C>T形成复合杂合关系。结合临床表现和家系特点,可判定例1 患儿的复合杂合变异为致病变异。例2 患儿ABCB 11基因的21 号外显子检测到1 个纯合错义变异c.2606 A>C,起源于父母,导致ABCB 11基因的第869 位密码子由编码谷氨酰胺变为编码脯氨酸(p.Gln869Pro)(图3)。检索数据库及文献发现,c.2606A>C为已知的致病变异。
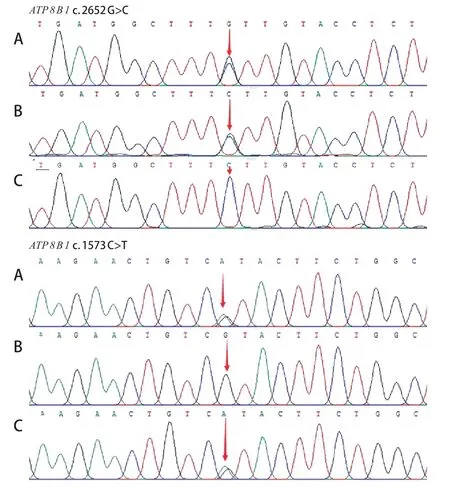
图2 例1 患儿及父母ATP8B1 基因测序图
2 讨论
PFIC 是一组以胆汁淤积为主要特征的遗传性疾病,主要表现为黄疸、皮肤瘙痒和肝功能异常,瘙痒是多数患者最明显的临床特征,其严重程度与黄疸程度不成正比。PFIC1,也称为拜勒病,由ATP8B1基因变异所致;PFIC 2,也称为胆汁酸盐输出泵疾病,由ABCB 11基因变异所致;PFIC 3,由ABCB 4基因变异所致,血生化特点为γ-谷氨酰转肽酶(GGT)持续升高。随着基因诊断的不断进展,近几年PFIC 4、PFIC 5、PFIC 6 首次被报道,分别由TJP 2、NR 1 H 4、MYO 5 B基因变异所致[3]。
PFIC 1 由基因ATP 8 B 1(染色体18 q 21)的纯合或复合杂合变异引起。ATP 8 B 1起脂质翻转酶的作用,将磷脂酰丝氨酸和磷脂酰乙醇胺从胞浆带入膜小管小叶胞质,还可维持质膜内叶和外叶之间的不对称性,对肝细胞具有保护作用[4]。ATP 8 B 1在多个器官表达,包括肝脏、胰腺、肾脏和小肠,其中表达最多的是小肠。因此PFIC的临床表现还可有肝外表现,如慢性腹泻、身材矮小、耳聋、脾大、胰腺炎等[5]。例1 患儿存在皮肤瘙痒、黄疸、慢性腹泻、脾大,基因检测发现ATP8B1基因存在c.2652G>C和c.1573C>T复合杂合变异,其中c.2652G>C尚未见收录,c.1573C>T已有文献报道。依据生物信息学分析及ACMG 指南,c.2652G>C可能为致病变异,结合临床表现及基因检测结果,例1可诊断为PFIC1。
PFIC2由基因ABCB11(染色体2q24)变异引起,这是由BSEP 缺陷引起的PFIC 形式。对于常染色体隐性遗传疾病来说,单个致病性变异的携带者并不会发展为患者,只有纯合致病性变异或复合杂合致病性变异才会导致疾病发生。BSEP 是肝脏特异的三磷酸腺苷结合盒转运蛋白超家族成员,是胆盐的主要转运体,在胆汁的肝肠循环中起关键作用[6-7]。BSEP 缺乏会导致新生儿胆汁淤积性黄疸和瘙痒;GGT 无异常,AST 和/或ALT 明显升高,血胆汁酸浓度升高;肝组织病理显示胆汁主要淤积在肝细胞内,而不是在毛细胆管内;光镜下表现为明显肝多核巨细胞形成,肝细胞胆汁淤积,伴有胆管增生、门静脉纤维化和肝硬化,髓外造血等;电镜下胆汁呈细丝状、细颗粒状或无定形状,微绒毛缺失[8]。例2 患儿存在黄疸、皮肤瘙痒,且GGT 无异常、AST 升高、总胆汁酸升高,肝脏病理结果符合上述表现,且基因检测发现ABCB11基因存在c.2606A>C纯合错义变异,多种生物信息预测软件预测此变异会对基因或基因产物造成有害影响,故可诊断为PFIC2。
PFIC目前尚无特异的治疗方法,治疗目标为控制黄疸和瘙痒,延缓胆汁淤积对肝脏的进一步损伤。非手术治疗方案主要是使用熊去氧胆酸、利福平和补充脂溶性维生素[6]。熊去氧胆酸可以促进胆汁排出,缓解胆汁淤积,有助于保护肝细胞。利福平可以诱导药物代谢酶(CYP 3 A 4)的表达,增加胆红素结合物和排泄量,缓解瘙痒症状[9]。另一种非手术干预是鼻胆管引流术,建立暂时性的胆汁转移,不过需要考虑与手术相关的胰腺炎风险。但是,这些非手术方法并非对所有患者都起效,即使有效通常也不能完全缓解瘙痒。因此部分患者需要手术治疗,例如部分胆汁外分流术或部分胆汁内分流术,以降低循环胆汁酸浓度。最终,患者通常因为肝衰竭和顽固性瘙痒进行肝移植手术[5]。需要注意的是,由于ATP8 B1的表达不仅限于肝脏,即使肝移植后,肝外表现诸如胰腺炎、身材矮小和听力下降等也将持续存在,并且可能引起腹泻加剧。此外,移植后常会发生严重的肝脂肪变性,逐渐发展为肝硬化,需要再次移植。在一些严重的ABCB 11缺陷患者中,由于形成了针对BSEP的自身抗体,肝移植后会再次出现胆汁淤积,可能也需要进行再次肝移植[10]。研究显示,肝细胞移植、变异特异性疗法以及靶向疗法可能为PFIC患者提供更多的治疗方法,同时精确的分子诊断能够为患者个性化治疗提供更可靠的依据[6]。本组2例患儿目前均接受熊去氧胆酸治疗,间断补充脂溶性维生素,黄疸、皮肤瘙痒较前明显缓解;目前肝功能正常,未出现肝硬化,需要进一步观察预后。
综上所述,PFIC临床表现为不同程度的黄疸及皮肤瘙痒,疾病相关基因检测对于疾病早期诊断、早期干预治疗进而提高患儿的预后及生存质量有重要价值。本研究发现1个未见报道的ATP8B1基因变异,丰富了基因变异数据库,为今后的研究提供了临床资料。
猜你喜欢
基层中医药(2022年7期)2022-11-17
现代临床医学(2022年4期)2022-09-29
现代仪器与医疗(2022年3期)2022-08-12
肝博士(2022年3期)2022-06-30
保健医苑(2022年5期)2022-06-10
医学前沿(2021年2期)2021-09-10
珠江水运(2021年15期)2021-08-29
昆明医科大学学报(2021年5期)2021-07-22
中西医结合肝病杂志(2020年2期)2020-10-27
黄河黄土黄种人(2017年11期)2017-11-27
