
抑制p38/ATF2信号通路对氯化锂-匹罗卡品致癫痫大鼠海马神经元损伤的保护作用研究
2021-07-05 08:05韩仲谋张其梅
巴楚医学 2021年2期
韩仲谋 夏 杰 陈 静 张其梅
(三峡大学 第一临床医学院[宜昌市中心人民医院] 神经内科, 湖北 宜昌 443003)
癫痫是一种常见的神经系统慢性发作性疾病。我国流行病学调查资料显示,癫痫患病率为0.90‰~4.80‰,农村年发病率为25/10万,城市为35/10万[1]。由于癫痫的发病机理极为复杂,至今尚未完全明确,故其治疗缺乏突破性进展。丝裂原活化蛋白激酶(mitogen activated protein kinases,MAPKs)是目前发现的最重要信号转导蛋白之一,它在细胞增殖、分化、生长、死亡过程中起着关键作用[2]。研究表明,MAPK在癫痫患者中表达上调,提示该蛋白可能具有致病作用[3]。p38/活化转录因子2(activated transcription factor 2,ATF2)轴可促进中枢神经系统小胶质细胞的炎症活化,从而加重脑内神经元损伤,成为治疗中枢神经系统疾病的潜在靶点[4, 5]。本研究以氯化锂-匹罗卡品大鼠癫痫模型为基础,通过观察海马神经元中MAPK家族成员p38及其下游转录因子ATF2的活化规律,探讨p38信号通路对海马癫痫神经元损伤的影响及相应的神经保护调节机制。
1 材料与方法
1.1 试剂和设备
主要试剂和设备:匹罗卡品、氯化锂购于美国Alexis公司,抑制剂SB203580、二甲亚枫(dimethyl sulfoxide,DMSO)购于美国Sigma公司,鼠源p38抗体、鼠源p-p38抗体购于美国Santa Cruz Biotechnology公司,兔源ATF2抗体、兔源p-ATF2抗体购于美国Abcam公司,辣根过氧化物酶标记二抗购于北京中杉金桥生物技术有限公司,BCA蛋白定量试剂盒、SDS-PAGE凝胶试剂盒购于西安晶彩生物科技有限公司。垂直电泳槽、凝胶成像系统购于美国Bio-Rad公司,PVDF膜购于美国MILLIPORE公司,BX43型双目生物摄像显微镜购于日本Olympus公司。
1.2 实验动物
实验动物:清洁级成年雄性SD大鼠36只,单体质量为(200±20)g,购自三峡大学医学院动物实验中心,动物合格证号:No.42010200000426;No.42010200000432。
36只大鼠随机分为4组:假手术组、癫痫组、抑制剂组、对照组,每组9只,所有大鼠均提前行侧脑室置管。假手术组:腹腔注射生理盐水,侧脑室注射生理盐水10 μL。除假手术组外,其余3组均需构建癫痫持续状态(status epilepticus,SE)大鼠模型。癫痫组:清醒状态下,腹腔注射氯化锂3 meq/kg,18 h后,腹腔注射盐酸匹罗卡品50 mg/kg,注射匹罗卡品前30 min侧脑室注射生理盐水10 μL。抑制剂组:p38抑制剂SB203580溶于10 g/L DMSO,取10 μL侧脑室注射。对照组:侧脑室注射等体积10 g/L DMSO。按经典的Racine癫痫发作行为标准观察和记录。从Ⅳ级发作开始计时,分别在30 min、2 h、6 h开始采集标本。
1.3 侧脑室置管及注射
大鼠以氨基甲酸乙酯腹腔麻醉,俯卧位固定,颅顶去毛消毒,沿矢状缝做一长约1 cm切口,分离筋膜、骨膜,暴露前囟,据Paxinox立体定位图谱进行定位,坐标为:前囟前0.4 mm,正中线右旁开1.5 mm,硬膜下2.2 mm,以牙科钻在定位点钻一直径约1 mm圆孔至有落空感,或有少量清亮脑脊液渗出,将注射内管连接导管后垂直插入孔内,将微量注射器与PE连接管连接,10 min内匀速注射10 μL,留针5 min,关闭伤口。
1.4 实验动物标本取材
在相应时间点, 5%水合氯醛腹腔注射麻醉大鼠,4%冷多聚甲醛磷酸盐缓冲液灌注固定后,根据Paxinox立体定位图谱取海马组织,经冠面切开分成相等的两份,一份装入EP管立即-80℃保存,用于Western blot检测,另一份石蜡包埋冠状切片用于HE染色。
1.5 Western blot检测
取-80℃保存的大鼠海马组织,加入PBS溶液,微离心弃上清液,重复1~2次直到上清液清亮,加入RIPA裂解液匀浆、离心,分装上清液-80℃保存。按BCA试剂盒要求测定蛋白浓度。每组取50 μL 样品,加入上样缓冲液,加热至95℃维持5 min使蛋白变性,而后行10%十二烷基硫酸钠聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE),将纯化后的蛋白转移到PVDF膜上,经TBST洗膜后5%脱脂奶粉封闭2 h,分别加入鼠源p38(1∶500稀释)、鼠源p-p38(1∶1 000)、兔源ATF2(1∶1 000)、兔源p-ATF2(1∶1 000)一抗,4℃孵育过夜。经TBST 洗膜3次,每次5 min,加入辣根过氧化物酶标记的二抗,在脱色摇床上于室温下反应1 h。TBST洗膜,加入ECL发光液。利用凝胶成像系统和Image Lab软件对目标蛋白条带进行分析。
1.6 组织病理学观察
标本常规制成石蜡切片,HE染色,光镜下观察癫痫持续不同时间的海马CA1区、CA3区、齿状回颗粒细胞组织病理学变化。
1.7 统计学处理
采用SPSS 19.0统计软件进行数据分析,计量资料以均数±标准差表示,组间比较采用单因素方差分析,不同时间点、不同组别比较采用q检验,P<0.05为差异有统计学意义。
2 结果
2.1 匹罗卡品致癫痫持续状态大鼠模型的建立
所有大鼠均由面部抽搐前肢肌阵挛发作,随后发展为全身性阵挛发作,可有后肢站立;最后发展为持续性全身性强直-阵挛,同时伴后肢站立及跌倒发作,符合Racine分级V级发作, SE大鼠造模成功。
2.2 癫痫大鼠海马CA1区神经元形态学变化
光镜下观察可见,假手术组CA1神经元整齐排列,细胞形态完整,胞核呈圆形,染色质分布均匀,核仁可见,较清晰。癫痫组CA1区神经元结构紊乱,神经元核固缩,核仁未见,胞体缩小变形,胞质浓缩深染,尼氏小体消失,典型的多角形变为三角形,又称为“红色神经元”。在各时间点,抑制剂组CA1区神经元损伤程度均明显轻于癫痫组(见图1)。
2.3 各组大鼠海马脑组织中p38、p-p38、ATF2、p-ATF2蛋白表达
与假手术组相比,癫痫组p38和ATF2蛋白表达量均无明显差异(均P>0.05),p-p38和p-ATF2蛋白表达量均明显增高(均P<0.01),p-p38/p38比值和p-ATF2/ATF2比值均明显增高(均P<0.01),6 h时达峰值。与癫痫组和对照组相比,抑制剂组p38和ATF2蛋白表达量均无明显差异(均P>0.05),p-p38和p-ATF2蛋白表达量均明显降低(均P<0.01),p-p38/p38比值和p-ATF2/ATF2比值均明显降低(均P<0.01)(见图2)。

注:各组海马CA1区HE染色(×200)图1 癫痫大鼠海马CA1区神经元形态
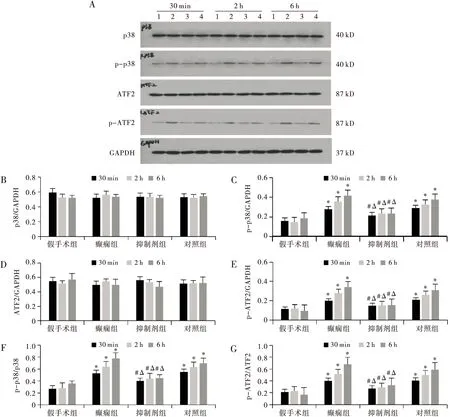
注:A:各组大鼠海马脑组织中p38、p-p38、ATF2、p-ATF2蛋白表达western blot条带图, 1为假手术组,2为癫痫组,3为抑制剂组,4为对照组;B~G:各组大鼠海马脑组织中p38、p-p38、ATF2、p-ATF2、p-p38/p38、p-ATF2/ATF2相对表达柱状图。同一时间,与假手术组相比,*P<0.01;同一时间,与癫痫组相比,#P<0.01;同一时间,与对照组相比,ΔP<0.01图2 各组大鼠海马脑组织中p38、p-p38、ATF2、p-ATF2蛋白表达
3 讨论
本研究显示,抑制剂组与癫痫组相比,CA1区存在部分变性的神经元,较癫痫组数量减少,提示神经元受损减少。证实癫痫发生后,大鼠海马CA1区神经元出现凋亡[6],p38信号通路参与其发生发展过程,SB203580通过竞争性结合ATP结合位点来抑制p38MAPK活化,拮抗某些炎症因子的信号转导[7],进而减轻海马神经组织的损伤。
目前认为海马区病变与癫痫的发生发展紧密相关。海马损伤时会发生病理改变,这些改变主要包括神经元损伤、苔藓纤维出芽(mossy fiber sprouting,MFS)、胶质细胞增生[8]。海马神经元损伤和丢失是诱发MFS的必要因素[9]。癫痫时星形胶质细胞的功能被激活,生理上这些细胞的功能激活依赖于海马区神经元的脱失和微环境的改变,导致胶质细胞发生增生及慢性瘢痕形成[10]。癫痫发生时,海马苔藓纤维异常发芽为主要病理改变,这也是构成癫痫反复长期发作的原因之一。
p38是突触内含量最丰富的一种囊泡膜蛋白,和癫痫、帕金森病、阿尔茨海默病等密切相关,可以被炎性细胞因子和环境应激等刺激激活,涉及炎性和凋亡过程相关的病理变化[11,12]。本实验中发现,致痫后2 h,p38在海马CA1区磷酸化水平增加,考虑为伴随苔藓纤维出芽,且形成新突触或突触重构,改变了原有的局部环路,颗粒细胞发生出芽侧枝回返,出芽纤维穿过颗粒细胞层到达齿状回分子层,建立新的兴奋联系,级联信号反应不断放大,诱导癫痫复发[13]。
MAPK信号转导途径包括p38MAPK、JNK、ERK5、ERK1/2,广泛参与细胞的生长、分化、凋亡等病理生理过程[14]。多项研究证实,p38MAPK在急、慢性癫痫的致病过程中发挥重要作用[15-17]。本研究发现,癫痫后p38磷酸化水平明显增加,可能与p38MAPK信号转导通路激活促进神经元死亡有关。癫痫的大鼠在经过SB203580(p38MAPK抑制剂)预处理后,p38及ATF2的磷酸化活化均较癫痫组减弱。SB203580通透细胞,抑制p38MAPK通路,考虑与其抑制MAPKAP Kinase-2和 Kinase-3的激活、同时拮抗炎症因子白介素(interleukin,IL)-1β、肿瘤坏死因子(tumor necrosis factor,TNF)-α等相关[15]。
ATF2蛋白是AP-1家族成员之一,位于2q31染色体,ATF2的羧基端拉链结构域与其自身氨基端锌指结构结合形成同源二聚体,羧基端结构域与氨基端结构域之间互为抑制,使转录过程受到阻止[18]。当受到缺氧、炎性因子影响时,ATF2的Thr69/71位点可被PKC、SAPK、p38、MAPK、JNK磷酸化激活,该激活位点与c-jun或c-fos的CRE原件结合形成异源二聚体,启动转录元件,介导下游基因的表达,调控细胞的增殖、分化、凋亡等生物学过程[19]。p38通路被各种应激信号比如缺氧、炎症因子等内源性、外源性因素激活后,下游底物ATF-2、c-Jun发生磷酸化,磷酸化后的ATF-2与c-Jun启动子相结合,c-Jun的转录活性进一步提升,环氧化酶-2的表达增加,导致神经元损伤和凋亡[20]。
有研究指出,在癫痫发作的急性期、SE中均检测到p38信号通路的激活[21]。在前期的研究中我们也发现,JNK和p38信号转导通路在氯化锂-匹罗卡品致痫过程中被激活[22]。本研究提示,抑制p38/ATF2信号转导可改善SE诱导的海马神经元损伤,为揭示癫痫神经元损伤的分子机制及寻找癫痫防治靶点提供了有利线索。
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05
健康必读·下旬刊(2019年8期)2019-08-16
江苏农业科学(2017年14期)2017-10-10
天津农业科学(2017年8期)2017-08-11
饮食科学(2017年5期)2017-05-20
吉林农业(2017年5期)2017-05-13
家庭医药(2016年8期)2016-09-28
科技知识动漫(2016年6期)2016-06-24
江苏农业科学(2015年5期)2015-10-20
汽车观察(2009年1期)2009-02-18
