
11例抗神经元抗体阳性的自身免疫性脑炎的临床特征
2021-07-05 03:27张其梅
巴楚医学 2021年2期
郑 铮 张其梅 夏 杰
(三峡大学 第一临床医学院[宜昌市中心人民医院] 神经内科, 湖北 宜昌 443003)
自身免疫性脑炎(autoimmune encephalitis,AE)泛指一类由自身免疫机制介导的脑炎[1]。其中,由于神经元表面或突触蛋白受到相应自身抗体免疫攻击所致的脑炎是一类新型的AE,又称“狭义的AE”。该疾病在过去的十余年中引起了广泛的关注,它可以引起严重的临床表现,并可能导致死亡。这一大类新型AE主要通过体液免疫机制引起相对可逆的神经元功能障碍,免疫治疗效果良好[2-4]。尽早确诊及启动免疫治疗,可改善患者预后。因此,对该病的早期识别和诊断非常重要。本文对我科收治的11例抗神经元抗体阳性AE患者的临床资料进行分析,以总结AE的临床特征、治疗和结局,提高临床医生对该病的认识。
1 研究对象和方法
1.1 研究对象
本回顾性研究纳入了2018年1月~2020年3月在我院神经内科诊断的11例抗神经元抗体阳性的AE病例。所有患者均符合以下入选标准:①符合2017年中华医学会神经病学分会发布的《中国自身免疫性脑炎专家共识》诊断标准;②急性期接受一线免疫治疗,包括糖皮质激素和/或免疫球蛋白和/或血浆置换;③患者血清或脑脊液有1项或多项抗神经元抗体阳性;④通过门诊或电话随访时间超过6个月,并有完整的医疗记录。排除了可能的AE,但抗体检测阴性者以及其他可能的病因,如病毒性脑炎或心理疾病的患者。
1.2 研究方法
分析患者的临床资料,包括性别、起病年龄、首发症状、全部症状、合并症(肿瘤和/或其它疾病)、检验结果(脑脊液常规、生化、血清和/或脑脊液自身抗体)、影像学资料、脑电图表现、治疗方案及结局。采用改良的Rankin量表(modified Rankin Scale,mRS)对患者神经功能结局进行评价。所有患者的血和脑脊液AE抗体检测由武汉康圣环球医学检验所完成。
1.3 统计学方法
采用SPSS 23.0统计软件进行数据处理与分析。计数资料以相对数构成比(%)或率(%)表示。
2 结果
2.1 一般资料
11例患者中,男性7例,女性4例,发病年龄17~63岁;平均年龄(44.45±17.90)岁,中位年龄48岁。其中5例为N-甲基-D-天冬氨酸受体(N-methyl-D-aspartic acid receptor,NMDAR)抗体阳性(45.45%);5例为γ-氨基丁酸B受体(γ-aminobutyric acid B receptor,GABABR)抗体阳性(45.45%),其中1例合并抗Hu抗体阳性;1例为抗富含亮氨酸胶质瘤失活蛋白(leucine glioma inactivated protein 1,LGI1)抗体阳性(9.09%)(详见表1)。

表1 11例抗神经元抗体阳性AE患者的一般资料
2.2 临床表现
2.2.1 临床症状
所有患者均急性或亚急性起病。最初表现为癫痫发作6例(54.55%),是最常见的首发症状,其中1例(9.09%)患者合并有面-臂肌张力障碍发作(faciobrachial dystonia seizures,FBDS);精神行为异常或认知功能障碍5例(45.45%);有3例(27.27%)患者在疾病的初期有发热。在整个病程中,9例(81.82%)患者出现完全性脑病表现(精神异常+癫痫发作+认知障碍),7例(63.64%)患者出现意识水平下降或昏迷,2例(18.18%)患者出现低钠血症(详见表2)。

表2 11例抗神经元抗体阳性AE患者的临床表现
2.2.2 合并症情况
当患者被怀疑为AE时,进行了肿瘤筛查(胸部、腹部和盆腔的MRI和/或CT和/或超声)、血清肿瘤标记物检查。1例患者被发现有右肺肿瘤,经病理活检证实为小细胞肺癌(small cell lung cancer,SCLC),该患者血清及脑脊液抗GABABR抗体及抗Hu抗体均阳性。1例患者合并卵巢畸胎瘤,其血清及脑脊液抗NMDAR抗体呈阳性。其余患者在发病时及以后的随访中均未发现肿瘤(见表2)。
2.2.3 辅助检查结果
辅助检查结果详见表3。脑脊液检查中脑脊液白细胞增多标准为>5×106/L,脑脊液蛋白升高为>450 mg/L,脑脊液压力升高为>200 mmH2O。7例患者脑脊液白细胞计数升高,其中1例白细胞计数达166×106/L,其余6例患者白细胞计数均<50×106/L,白细胞增多全部以淋巴细胞为主;4例患者脑脊液蛋白升高,均为轻度升高,<600 mg/L;4例患者脑脊液压力升高,均<300 mmH2O。
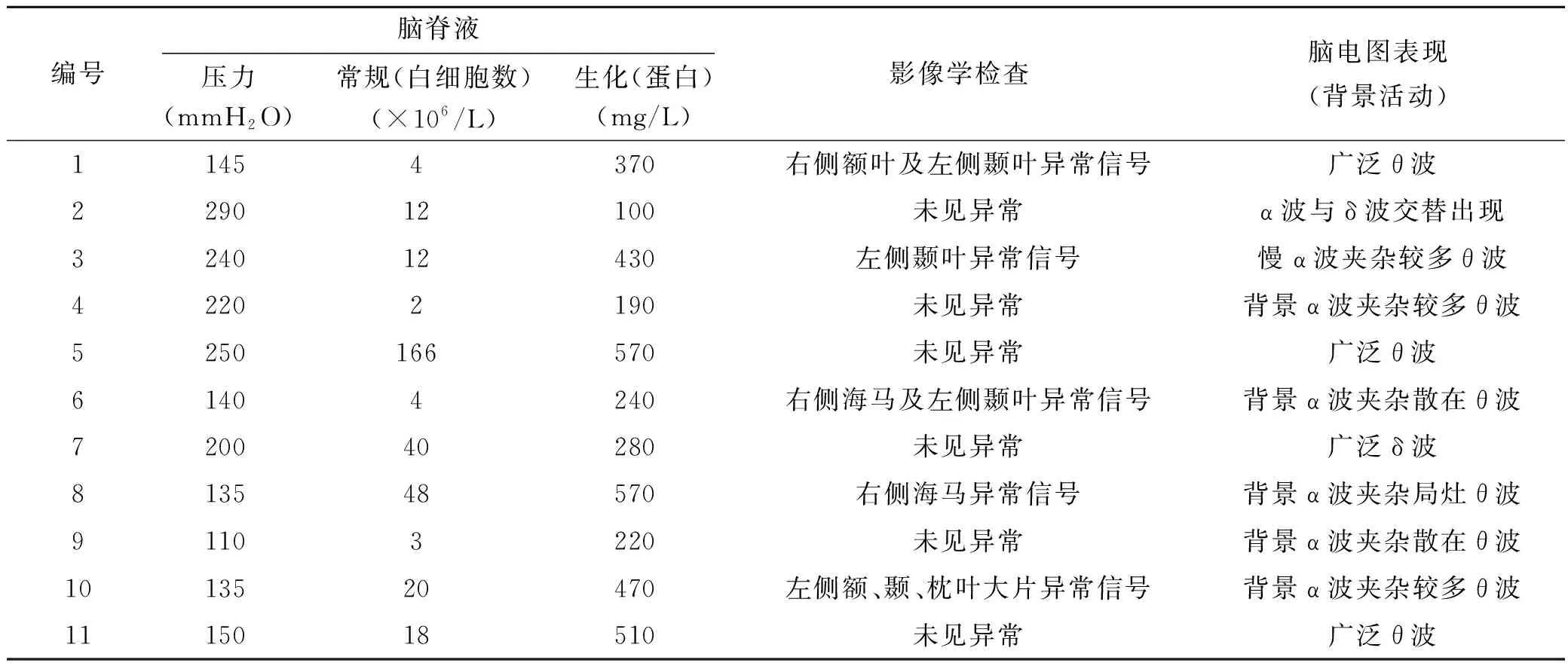
表3 11例抗神经元抗体阳性AE患者的辅助检查结果
所有患者均接受了至少1次头颅MRI平扫及增强扫描检查。脑MRI异常表现被定义为T2加权图像(T2-weighted image,T2WI)、液体衰减反转恢复(fluid-attenuated inversion-recovery,FLAIR)图像或弥散加权成像(diffusion weighted imaging,DWI)的高信号和/或T1加权图像(T1WI)的低信号。11例患者中6例脑MRI表现正常。剩下的5例表现异常:1例抗LGI1抗体脑炎患者病灶在右侧额叶及左侧颞叶;1例抗GABABR抗体脑炎病灶在左侧颞叶,且病灶面积大;1例抗NMDAR抗体脑炎病灶在右侧海马及左侧颞叶;1例抗GABABR+Hu抗体脑炎病灶在右侧海马;1例抗GABABR抗体脑炎病灶广泛分布于左侧额、颞、枕叶(见表3和图1)。
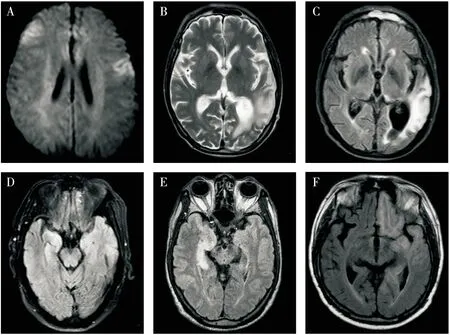
注:A:抗LGI1抗体脑炎右侧额叶及左侧颞叶病灶;B、C:抗GABABR抗体脑炎左侧颞叶大片病灶;D:抗NMDAR抗体脑炎右侧海马及左侧颞叶病灶;E:抗GABABR+Hu抗体脑炎右侧海马病灶:F:抗GABABR抗体脑炎广泛分布于左侧额、颞、枕叶的病灶图1 抗神经元抗体阳性AE患者的异常影像学表现
所有患者至少进行1次数字视频脑电图记录,脑电图记录至少30 min。脑电图数据分类如下:背景活动正常、广泛慢化、局灶慢化和极度δ刷(extreme delta brushes,EDB)。11例患者存在不同程度的慢波增多,1例患者表现为广泛δ波;3例患者表现为广泛θ波;1例患者表现为阵发α波与δ波交替出现;其余患者表现为背景α波中夹杂不同程度的慢波。
2.3 免疫治疗
所有患者在急性期或亚急性期均接受了一线免疫治疗(糖皮质激素、免疫球蛋白、血浆置换或这些治疗的任意组合)及对症治疗(抗癫痫、抗精神症状及合并症治疗),见表4。除1例患者合并糖尿病酮症酸中毒,仅给予醋酸泼尼松口服,其余患者全部给予大剂量甲泼尼龙静脉滴注后逐渐减量改为口服醋酸泼尼松。1例患者早期使用甲泼尼龙冲击治疗,后期合并消化道出血,停用口服醋酸泼尼松。4例患者接受了二线免疫治疗(利妥昔单抗),其中1例患者因出现不良反应而提前停药。2例患者接受了长程免疫治疗(吗替麦考酚酯),其中1例患者因不良反应明显,使用1次后停药。
2.4 结局
患者随访时间至少6月,根据mRS评分评价结果。评价标准:完全康复,mRS评分0分;轻度缺陷,mRS为1~2分;中度残疾,mRS为3分;严重缺陷,mRS为3~5分;或者死亡,mRS为6分。3例患者完全康复,3例患者轻度缺陷,3例患者中度残疾,2例患者死亡。1例抗NMDAR抗体脑炎合并卵巢畸胎瘤患者经免疫及对症治疗后脑炎症状好转,行畸胎瘤切除术,最终患者遗留轻度缺陷。病例8为抗GABABR+Hu抗体脑炎合并右肺小细胞肺癌,经免疫及对症治疗后mRS得分1分,患者转外科行肺癌切除术,术后合并重症感染、脓毒性休克死亡(详见表4)。

表4 免疫治疗方案和结局
3 讨论
AE的报道在近几年逐渐增多,随着临床医师对该疾病认识的增强,对更多的疑似患者进行了自身抗体的检测,因而出现了更多的确诊患者。根据不同的抗神经元抗体和相应的临床综合征,狭义的AE可分为3种主要类型:①抗NMDAR脑炎;②边缘性脑炎,包括抗LGI1抗体、抗GABABR抗体与抗α氨基-3-羟基-5-甲基-4-异噁唑丙酸受体抗体相关的脑炎;③其他AE综合征,包括莫旺综合征(Morvan′s syndrome)、抗GABAAR抗体相关脑炎、伴有强直与肌阵挛的进行性脑脊髓炎(progressive encephalomyelitis with rigidity and myoclonus,PERM)、抗多巴胺2型受体(D2R)抗体相关基底节脑炎、抗二肽基肽酶样蛋白(DPPX)抗体相关脑炎、抗IgLON5抗体相关脑病等[1]。本研究11例患者中,诊断了抗NMDAR脑炎、抗GABABR脑炎、抗LGI1脑炎3种AE。
抗NMDAR脑炎是最常见的AE[5],任何年龄段的人均可发病,年轻妇女和儿童为高发人群。在>45岁的患者中,男性更为常见[6]。本研究5例抗NMDAR脑炎患者中4例为女性,年龄为17~42岁,1例为男性,年龄48岁。研究已发现,抗NMDAR脑炎发生频率超过所有个体病毒性脑炎[7]。在相当数量的患者中,神经体征和症状都与肿瘤有关,最常见的是卵巢畸胎瘤,也有文献报道合并睾丸未成熟畸胎瘤和淋巴瘤[8,9]。但是该疾病与肿瘤的关联在儿童中并不常见。有研究报道,年龄在18岁以下的患者中有9%合并肿瘤,而年龄在18岁以上的患者中有27%合并肿瘤[10]。本研究5例抗NMDAR脑炎患者中1例合并卵巢畸胎瘤,为31岁女性。从临床角度来看,抗NMDAR脑炎的特征是精神症状、行为异常、意识下降、言语和运动障碍、自主神经功能障碍、中枢性通气不足、认知障碍和癫痫发作。EDB是一种特殊的脑电图模式,出现在30%的抗NMDAR脑炎患者[11]。35%的患者在发病时脑MRI异常,50%的患者可表现为晚期异常,主要为灰质和白质的非特异性高强度病变。罕见病例的病灶提示脱髓鞘病变与脱髓鞘综合征重叠,如与水通道蛋白-4抗体相关的视神经脊髓炎谱系疾病或与髓鞘少突胶质细胞糖蛋白抗体相关的脱髓鞘疾病并存[8]。本研究中5例抗NMDAR脑炎患者脑电图均未发现EDB,1例患者脑MRI异常,这与文献报道的数据存在差异,考虑与本研究样本数量较小及行脑电图、MRI检查疾病所处的时期有关。抗NMDAR脑炎最终的预后通常是好的。在一项涉及252名患者(包括1/3的儿童和青少年)的研究中,诊断AE后随访24个月,死亡率为9.5%,83%有良好的预后[8]。本研究5例患者中,除1例患者死亡,其余4例均预后良好,与文献报道相符。这例患者死亡原因考虑与病情严重、并发症多及免疫治疗不能耐受有关。
关于抗GABABR脑炎的临床表现,最早由Lancaster等[11]报道,他们发现GABABR抗体与伴有严重癫痫发作的边缘叶脑炎的进展有关。患者可出现小脑性共济失调、斜视性眼阵挛-肌阵挛和脑干受累[12-14]。一项11例成人患者的研究[15]显示,该病主要发生于男性(7/11),大约50%的病例与肿瘤相关(5/11),特点是认知能力下降(9/11)、癫痫(10/11)、精神和行为障碍(6/11)、不自主运动(4/11)、睡眠障碍(2/11)、听力减退(1/11)、意识障碍(4/11)和发热(3/11);在脑电图上,5例患者出现癫痫样放电;4例患者脑MRI显示海马区、海马旁回、颞叶、枕叶异常信号;8例患者一线治疗反应良好。抗GABABR脑炎合并肿瘤在男性患者中多见[15]。癫痫发作是最常见的首发症状,所有患者在病程中均表现为典型的边缘叶脑炎。无肿瘤患者对免疫抑制治疗的反应良好。抗GABABR脑炎的长期预后取决于潜在的恶性肿瘤[16]。本研究5例抗GABABR脑炎患者均对免疫抑制治疗有反应,1例患者合并抗Hu抗体阳性及右肺小细胞肺癌,经免疫及对症治疗后mRS得分1分,后转外科行肺癌切除术,术后合并重症感染、脓毒性休克死亡。再一次证实了合并的恶性肿瘤决定了抗GABABR脑炎的长期预后。
抗LGI1脑炎主要发生在60岁左右的男性,在成年人的发生率仅次于抗NMDAR脑炎[16],呈边缘叶脑炎的表现。癫痫发作和亚急性进行性记忆减退,行为和空间定位异常是这种疾病最常见的症状。在大约50%的病例中,最典型的癫痫发作是FBDS。FBDS是单侧面部和手臂(或腿)不自主的收缩,持续时间<3 s,一天可发生上百次,是抗LGI1脑炎的特征性表现。11%的抗LGI1脑炎患者可能会发现肿瘤,其中胸腺瘤和肺癌最常见[17]。约60%的病例存在低钠血症,约25%的患者发病时MRI正常,异常MRI主要表现为中颞叶T2高信号,与边缘叶脑炎一致,然而其长期评估也可能显示41%~95%的患者有海马萎缩或硬化[18]。在大多数情况下,抗LGI1脑炎对治疗的反应良好,免疫治疗开始早的患者更易获得良好的疗效。免疫治疗可迅速减少癫痫发作,但认知能力的提高可能较慢[18]。本研究中1例抗LGI1脑炎患者为56岁男性,出现了癫痫发作、FBDS、认知功能下降、低钠血症、MRI异常,经一线免疫治疗遗留轻度神经功能缺陷,与文献报道相符。
根据2017年中国专家共识,AE的免疫治疗包括一线免疫治疗(糖皮质激素和/或免疫球蛋白和/或血浆置换)、二线免疫治疗(利妥昔单抗/静脉用环磷酰胺)及长程免疫治疗(吗替麦考酚酯/硫唑嘌呤等)。患者在一线免疫10~14 d后仍未好转,应接受二线治疗[2,8]。对于复发与难治性病例,可应用吗替麦考酚酯等口服免疫抑制剂[1]。对于合并肿瘤者,需尽早手术切除相关肿瘤,减少AE复发[19]。本研究中5例患者一线免疫治疗疗效不佳,接受了二线免疫治疗和/或长程免疫治疗,最终有3例获得较好疗效。由此可见,根据指南及时升级免疫治疗有助于改善预后。但本研究样本量较小,部分病例随访时间不长,其结论难免存在一定偏差,今后应该扩大样本量,进行长期随访研究。
综上所述,对于急性或亚急性起病的精神异常、癫痫发作、认知障碍的患者要考虑到AE的可能,尽早确诊AE便于尽早采用适当的治疗干预,改善患者预后。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
健康博览(2022年5期)2022-05-24
现代临床医学(2022年1期)2022-02-12
家庭医药(2020年10期)2020-10-30
科教新报(2019年26期)2019-09-10
家庭科学·新健康(2019年1期)2019-03-06
大众健康(2018年10期)2018-10-22
华东师范大学学报(自然科学版)(2018年3期)2018-05-14
为了孩子(孕0~3岁)(2001年3期)2001-06-13
祝您健康(1998年9期)1998-12-25
