
神经元蜡样脂褐质沉积症3 例临床及基因变异分析
2021-05-24 02:16禚志红孔惠敏靳培娜王怀立
临床儿科杂志 2021年5期
王 瑶 禚志红 孔惠敏 靳培娜 方 敩 王怀立
郑州大学第一附属医院儿科(河南郑州 450000)
神经元蜡样脂褐质沉积症(neuronal ceroid lipofuscinosis,NCL),也被称为Batten 病,是儿童时期常见的神经退行性疾病,主要临床表现为认知和运动功能减退、癫痫发作、进行性视力丧失、早逝[1]。此前NCL 按发病年龄分为4 类:婴儿型、晚婴型、少年型和成年型。随着遗传基因和生化的最新研究进展,学者对NCL 以基因分层方式进行了新的分类,以便有效地进行临床管理及作为未来基因治疗的基础[2]。目前PPT 1、TPP 1、DNAJC 5、CLN 3、CLN 5、CLN 6、MFSD 8、CLN 8、CTSD、GRN、ATP 13 A 2、CTSF和KCDT 7基因变异被认为与NCL 发病相关[3]。组织病理学检查或基因检测可明确诊断[4]。本研究分析3个家系NCL患儿的临床特点,并进行遗传学分析,以提高临床医师对该病的认识。
1 临床资料
例1,女,3岁4个月,因认知、运动倒退1年余,抽搐4 个月就诊。患儿3~4 个月抬头,6 个月会坐,7 个月翻身,11 个月扶站,1 周岁可以独走,8 个月会无意识喊“妈妈”。患儿1 岁半时开始出现认知、运动逐渐倒退,现不能独坐、不会走路、不会说话,不会咀嚼。3岁1个月时出现抽搐,表现为双目左斜、双上肢屈曲、四肢快速抖动,手心、足心多汗,约1 小时缓解。予服用左乙拉西坦、托吡酯,仍有发作,4、5 次/d。患儿系G2P2,足月剖宫产,出生体质量3.0 kg。患儿生后有新生儿高胆红素血症,予以蓝光照射治疗。母孕期体健,无有毒有害物质接触史。父母体健,非近亲结婚。有1个哥哥8岁,1岁半前发育正常,会独走、会说间断的单词,之后出现认知及运动倒退,现不会翻身、不能站立、不会说话、不会咀嚼;3 岁时开始出现抽搐,表现为双眼上翻、牙关紧闭、口吐白沫、四肢强直抖动,持续约半小时缓解;予以左乙拉西坦、复方苯巴比妥口服,抽搐较前有缓解;现间隔5~6 天发作1 次,表现为四肢快速抖动。体格检查:神清,竖头不稳,不能独坐,不会走路,双肺呼吸音清,心律不齐,四肢肌张力可,病理反射阴性。实验室检查:血常规、血生化、乳酸及血氨无异常。脑电图(EEG)正常。双眼彩超示右眼玻璃体轻浑,视神经回声前方可见小凹陷;左眼玻璃体内未见明显异常回声(图1)。头颅磁共振成像(MRI)示幕上脑室系统扩张,双侧大脑半球脑沟及脑裂可见增宽加深,考虑轻度脑萎缩(图2)。

图1 先证者1 双眼彩超报告

图2 患儿头颅MRI 表现
例2,女,5 岁2 个月,因发育倒退3 年余就诊。患儿3个月抬头,6~7个月会坐、翻身,12个月扶站,1岁2个月独走,1岁时会叫“爸爸、妈妈”。1岁半走路易摔跤,2岁半不能走路,2岁后不再说话,2岁左右咀嚼困难;现不能独坐,不会说话,流质饮食。患儿系G2P2,足月顺产,出生体质量3.3 kg。生后无窒息、抢救史。母孕期体健,无有毒有害物质接触史。父母体健,非近亲结婚。有1 个哥哥,1 岁半前发育正常,会独走、叫“爸爸、妈妈”;之后出现认知及运动倒退,现已去世。体格检查:神清,头可居中,不能独坐,不能站立,不认人;心肺腹无异常,足背屈角大,病理反射阴性。实验室检查:血常规、血生化、乳酸及血氨无异常。头颅MRI示额叶脑白质中心在T2WI呈低信号,额颞叶周围脑白质T2WI不明显;侧脑室前角旁及后角旁脑白质纠集;胼胝体偏薄;脑白质髓鞘化过程落后,脑沟裂增宽,可疑轻度脑萎缩,脑发育不良(图2)。
例3,女,6岁5个月,因发育倒退、抽搐1年余就诊。患儿3个月抬头,6~7个月会坐、翻身,1岁2个月会扶站,1岁5个月会独走,1岁2个月时会叫“爸爸、妈妈”;自幼智力稍差于同龄儿,但可正常上学。患儿于5岁3个月开始出现认知、运动的逐渐倒退,现可自行走路,但双腿发软;吐字不清、不会写字、不认字;同时伴抽搐,表现为双目上翻、眨眼、咂嘴、四肢强直,无口吐白沫、大小便失禁等症状,持续约1分钟缓解,缓解后嗜睡;未给予特殊治疗。6岁时再次出现抽搐,表现同前,给予左乙拉西坦口服。3个月前漏服药物后出现抽搐1 次,表现为突然摔倒、呼之不应、双眼半闭、四肢强直。现患儿规律服药,未再发作。患儿系G2P2,足月顺产,出生体质量4.0 kg。生后无窒息、抢救史。母孕期体健,无有毒有害物质接触史。父母体健,非近亲结婚。有1个姐姐14岁,体健。体格检查:智力落后,不认字,不能写字,不能准确计算10 以内加减法,吐字不清,视力下降,在1m外不能辨认家人,心肺腹正常,四肢肌张力可,病理反射阴性。实验室检查:血常规、血生化、乳酸及血氨无异常。视频脑电图示儿童异常脑电图;背景活动偏慢,清醒期广泛性或额区为主的慢波阵发,睡眠期左侧前额区或双侧枕区为主的尖慢波发放,监测中记录到发作期图形。视觉诱发电位示双眼P100潜伏时延迟,波形分化差。头颅MRI示小脑体积减少,小脑脑沟增宽,枕大池增大,提示小脑萎缩(图2)。
为进一步明确诊断,经医院伦理委员会批准及家属知情同意,对3 例患儿及其家属行全外显子基因检测。采用EDTA 抗凝试管,抽取先证者及家属外周血各2 mL,QIAGEN 基因组DNA 提取试剂盒(QIAamp DNA blood Minikit)提取外周血白细胞基因组DNA,利用NovaSeq 6000基因测序仪,将获得的质控合格的文库进行高通量测序,并根据基因检测结果进行Sanger 测序分析。结果显示,例1 的PPT 1基因存在复合杂合变异,c.124+1 G>A 和c.413 C>T,其中c.124+1G>A遗传自父亲,编码区第124号核苷酸后内含子中第1 位核苷酸由G 变为A;c.413 C>T遗传自母亲,导致编译第138 号氨基酸由丝氨酸变为亮氨酸(p.Ser138Leu),见图3。c.124+1G>A发生在splicing 区域,导致蛋白质功能改变;HGMD 数据库有此变异致病的报道。c.413C>T,蛋白功能预测结果为有害,疑似致病性变异;HGMD数据库有此变异致病的报道。结合临床表现和家系特点,可判定该复合杂合变异为致病变异。例2 及其哥哥的PPT 1基因存在复合杂合变异,c.181 C>T 和c.536+1 G>A,其中c.181 C>T 遗传自父亲,导致编译第61 号氨基酸Arg的密码子变为终止密码子(p.Arg61Ter),使肽链合成提前终止;c.536+1 G>A 遗传自母亲,编码区第536号核苷酸后内含子中第1位核苷酸由G变为A(图4)。两个变异位点分别在Clinvar和HGMD数据库中有致病报道。结合临床表现及检查结果,判定为致病变异。例3 的CLN 8基因存在复合杂合变异,c.768 G>T 和c.209G>T,其中c.768G>T遗传自父亲,导致编译第256号氨基酸由谷氨酰胺变为组氨酸(p.Gln256His);c.209 G>T 遗传自母亲,导致编译第70 号氨基酸由精氨酸变为亮氨酸(p.Arg 70 Leu),见图5。两变异位点均发生在CLN 8同源功能结构域,为中等性致病;c.768 G>T 和c.209 G>T 在千人基因组以及 HGMD 数据库中未收录。但结合临床表现、检查结果及家系特点,可判定该复合杂合变异为致病变异。
2 讨论
NCL是一组单基因遗传性退行性疾病,表现为神经元细胞内发现腊样脂褐质沉积、认知运动衰退、癫痫及视网膜病变。虽然所有的NCL临床和病理学都表现出相似性,但每一种类型都具有独特的病理生理特征。NCL 的超微结构可表现为颗粒状嗜锇物质、线样体、曲线体、指纹体结构[4]。随着对不同类型NCL 的深入研究,其表型谱也不断扩大。即使有相同基因变异的患者,临床表现也可能不同[1]。
CLN1基因位于染色体1p32,编码棕榈酰蛋白硫代酯酶1(palmitoyl protein thioesterase 1,PPT1)[4]。PPT1是一种可溶性溶酶体蛋白,参加多种细胞过程,包括凋亡、内吞、囊泡转运、突触功能和细胞内信号转导,其功能障碍可多种途径引起神经元凋亡。研究显示,PPT 1 也存在于溶酶体外,并不完全与溶酶体结合,表明其疾病可能并不仅仅是由于异常的储存所 致[5-6]。CLN8基因位于8p23.3,是一种非糖化跨膜蛋白,含有286个氨基酸,参与脂质的生物合成、转运和代谢[7]。
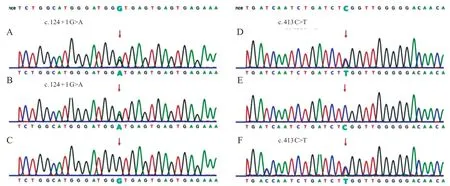
图3 先证者1 及其父母PPT1 基因测序图
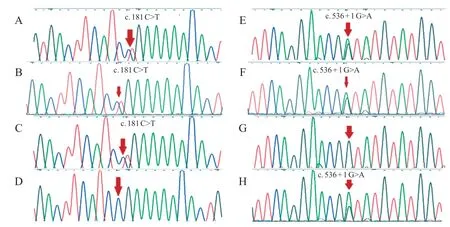
图4 先证者2 及其父母、哥哥PPT1 基因测序图
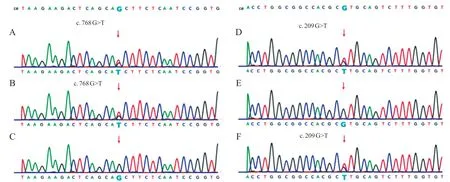
图5 先证者3 及其父母CLN8 基因测序图
典型婴儿型(6~24个月发病) 和晚婴型(2~5 岁发病)表现的第一症状是精神运动发育减慢,随后迅速停滞,然后是后天获得的认知和运动能力下降及癫痫发作,最后是视力丧失;而少年型(5~7岁发病)通常是视觉丧失,其次是精神和行为改变,最后在青春期早期丧失运动技能和癫痫发作[8]。本组患儿中,例1 和例2 家系中的4 例患者的发病年龄均在婴儿早期,临床表现均有认知和运动的倒退;例2 家系中后期伴有癫痫发作,符合婴儿型的临床表现。例3 的发病年龄较晚,临床表现为癫痫发作、认知和运动倒退及视力下降,考虑为晚婴型。文献报道,NCL 患儿癫痫主要发作形式为肌阵挛,可出现不典型失神发作、全面-强直阵挛发作或多种发作形式并存[9]。研究显示,CLN 1和CLN 8型患儿在疾病后期癫痫发作的频率会逐渐下降,但进行性痴呆和运动障碍继续 存在[7,10]。本研究中,例2 为肌阵挛癫痫,其哥哥的癫痫发作形式为多种形式并存;例3 为局灶强直阵挛发作,与文献报道相符[9]。基因检测显示,例1 为PPT 1基因c.124+1 G>A 和c.413 C>T 复合杂合变异。其中c.124+1 G>A 发生在splicing 区域,导致蛋白质功能改变。依据ACMG指南,具有极强的致病性。c.413C>T经相关蛋白功能分析软件预测为有害变异,判定为可疑致病变异。例2 为PPT 1基因c.181 C>T和c.536+1 G>A 复合杂合变异,2 个变异位点均可使蛋白质功能受到影响。结合临床表现及辅助检测,例1 和例2 均可诊断为NCL 1 型。例3 为CLN 8基因c.768 G>T 和c.209 G>T 复合杂合变异,2 个变异位点均未在Clinval 及HGMD 数据库中收录。生物信息学分析显示2 个变异位点对基因或基因产物均产生有害影响,结合临床表现及家系特点,可诊断为 NCL8型。
NCL患儿头颅MRI几乎都表现异常,主要包括弥漫性脑萎缩、白质脑病、皮质变薄和丘脑T2加权低强度。其中丘脑T2加权图像出现低强度最常见于婴儿,其次是青少年和晚期婴儿[11]。3例患儿的头颅MRI都表现有脑萎缩。随着病程进展,NCL患儿的脑萎缩程度会进行性加重,后期还需不断检测其变化。NCL 患儿EEG背景多为弥散性慢波,痫样放电可为局灶性和/ 或广泛性[9]。例1因未做长程EEG,不能提供有效信息。例3 的EEG 提示背景活动偏慢,广泛性痫样放电,与报道相符[9]。
NCL现尚无特异性的治疗手段。对于癫痫发作的患儿,可给予抗癫痫药物治疗,改善症状。新的治疗方法,如酶替代疗法、基因治疗、药理学方法、免疫调节疗法及神经干细胞移植疗法均处于临床试验中。目前脑室内注射酶替代疗法治疗CLN2是美国食品和药物管理局和欧洲药品管理局批准的第一种治疗方法,但预计需终生治疗[12-13]。
综上所述,PPT 1基因变异可呈现不同的临床表现,即使是同一家系具有相同变异点的个体临床表现也可各不相同,体现其表型异质性。疾病相关的基因检测有助于临床早期诊断,并进行遗传咨询。另外,在本组家系中发现2 个未见报道的CLN 8基因变异位点,丰富了基因变异数据库。随着分子生物学研究的不断进展,将有利于优生优育及提供有效的治疗 手段。
猜你喜欢
保健与生活(2022年9期)2022-05-06
健康博览(2021年7期)2021-08-16
保健与生活(2020年11期)2020-06-23
健康必读·下旬刊(2019年8期)2019-08-16
考试周刊(2017年26期)2017-12-12
校园英语·下旬(2017年7期)2017-07-14
饮食科学(2017年5期)2017-05-20
科技视界(2016年27期)2017-03-14
家庭科学·新健康(2016年9期)2016-10-25
家庭医药(2016年8期)2016-09-28
