
Wiskott-Aldrich 综合征患儿异基因造血干细胞移植后EB 病毒 相关淋巴组织增殖性疾病1 例报告并文献复习
2021-05-24 02:16习必鑫罗成娟
临床儿科杂志 2021年5期
习必鑫 陈 静 罗成娟
1.华中科技大学同济医学院附属同济医院儿童血液科(湖北武汉 430030);2.上海交通大学 医学院附属上海儿童医学中心血液肿瘤科(上海 200127)
关键字:Wiskott-Aldrich综合征;EB病毒;高危因素;治疗
Wiskott-Aldrich综合征(Wiskott-Aldrich syndrome,WAS)是一种罕见的X-连锁隐性遗传免疫缺陷病,根据其临床表型分为3种:WAS Ⅰ型(OMIM:301000)、X 连锁重型先天性中性粒细胞减少症(OMIM:300299)、血小板减少症Ⅰ型(OMIM:313900)。其中WAS Ⅰ型最常见,主要临床表现为血小板减少、湿疹、反复感染和频繁出血等[1],结合全外显子基因检测可以确诊。目前异基因造血干细胞移植(allogeneic hematopoietic stem cell transplantation,allo-HSCT)是唯一被证实可以治愈WAS的治疗方式。但是WAS患儿HSCT前的预处理方案中通常含有兔抗人胸腺细胞免疫球蛋白(anti-thymocyte globulin,ATG)治疗,而ATG又是发生移植后淋巴增殖性疾病(post-transplant lymphoproliferative disorder,PTLD)的独立高危因素之一。PTLD是实体器官移植(solid organ transplant,SOT)或造血干细胞移植(hematopoietic stem cell transplantation,HSCT)后患儿由于免疫系统过度抑制而诱发的一类淋巴组织良性增生甚至恶性增殖的疾病,是儿童移植后的严重并发症之一。儿童PTLD 的发生与移植后EB病毒(EBV)感染及免疫系统重建程度密切相关。本研究通过分析1例WAS合并卡斯尔曼病(Castleman disease)患儿allo-HSCT后发生EBV相关PTLD的临床诊疗资料,探讨儿童EBV-PTLD的高危因素及诊疗进展。
1 临床资料
患儿,男,G1P1,2006年11月足月剖宫产出生,出生体质量4 kg,出生后一般情况可。患儿8 个月大时体检发现外周血血小板20×109/L,遂在当地医院行骨髓穿刺检查示巨核细胞成熟差(全片巨核细胞20个),诊断免疫性血小板减少症,给予患儿输注丙种球蛋白及激素治疗后血小板上升,病情好转。近10余年间,患儿长期口服激素,并间断输注丙种球蛋白治疗,血小板一直波动于(20~40)×109/L。2018年1月(患儿11 岁2 个月),发现患儿左侧颈部淋巴结肿大,未予特殊处理。2018年12月(12岁),患儿双侧颈部可见大量肿大淋巴结,遂于外院就诊行淋巴结活检。病理检查报告示:Castleman 病(透明血管型);同时外送静脉血全外显子基因检测结果示患儿携带WAS基因c.961C>T(p.R321X)的半合子变异,考虑诊断为WAS合并Castleman病,建议患儿行HSCT治疗。2019年9月18日(13岁8个月),患儿入院行PV方案化疗(强的松45 mg/m2,1天1次;长春新碱1.5 mg/m2,1周1次),共2周,化疗后患儿双侧颈部肿大淋巴结明显消退。2019年10月初患儿获骨髓库HLA 配型10/10位点全相合供体,入院行allo-HSCT。
Allo-HSCT方案:①预处理,移植前(-9~-6天)白消安 0.8 mg/(kg·次),q 6 h×4 天;移植前(-5~ -2天),环磷酰胺50 mg/(kg·次),qd ×4天;移植前(-4~-2天)即复宁2.5 mg/(kg·次),qd ×3天。②骨髓库(HLA 配型10/10位点相合)外周血造血干细胞输注,移植当天(+0天)共输注CD34+细胞 5.1×106/kg,单个核细胞(ANC)17.6×108/kg。③急性移植物抗宿主病(graft versus-host disease,GVHD)预防,环孢素A(移植前1天开始)+短程甲氨蝶呤[MTX,移植后+1天,15 mg/(m2·次);+3天,10 mg/(m2·次);+6天,10 mg/(m2·次),共3次]。
移植后+6天,患儿出现肉眼血尿,遂将环孢素A口服剂量减至100 mg/d,静脉血环孢素药物浓度稳定于150~200 ng/mL之间,+12天患儿出血性膀胱炎好转。
移植后+1 2 天患儿外周血中性粒细胞计数≥0.5×109/L,中性粒细胞植入。移植后+14天外周血血小板≥20×109/L,血小板植入。移植后+16 天查外周血短串联重复序列聚合酶链反应(STR-PCR),99.4%为供者型。
移植后+21天,患儿出现频繁呕吐、剧烈腹痛,伴每天数10次墨绿色稀水样便,排除感染后,给予患儿口服吗替麦考酚酯片(MMF)1.0 g/d,呕吐、腹痛稍缓解,但每天仍有数10次稀水样便。遂于+29天给予患儿甲基泼尼松龙30 mg/d静滴,呕吐、腹痛、腹泻症状明显好转,+31天患儿消化道GVHD症状消失。
移植后+30 天,患儿血常规结果示白细胞计数5.79×109/L,中性粒细胞计数3.45×109/L,血红蛋白102 g/L,血小板计数89×109/L;环孢素A浓度227.3 ng/mL;血糖、肝肾功能无异常,电解质无异常,巨细胞病毒(CMV)-DNA阴性,EBV-DNA阴性。
移植后+33天,患儿外周血EBV-DNA为6×104拷贝/mL,无发热、淋巴结肿大等症状;予阿昔洛韦+膦甲酸钠抗病毒治疗,并逐渐减抗排异药(+52 天停用甲基泼尼松龙、+48 天停用MMF)。移植后+42天,患儿外周血T、B、NK病毒核酸分选:EBV-B DNA 1.39×106拷贝/mL,EBV-T DNA 1.35×105拷贝/mL,EBV-NK DNA 1.71×102拷贝/mL;+43天给予患儿第1次利妥昔单抗(375 mg/ m2)治疗。移植后+46天,患儿颈部出现肿大的淋巴结,颈部B超检查结果示右侧颈部淋巴结异常肿大,其中较大者23 mm×15 mm。+47 天,给予患儿第2 次美罗华治疗。+49 天,患儿右侧腋窝淋巴结可触及明显肿大。移植后+52 天,患儿右侧颈部肿大淋巴结病理检查结果示EBV 编码的小RNA(EBER)(+),EBV相关弥漫大B细胞淋巴瘤,浆母细胞表型(见图1)。移植后+57天、+64天,分别给予患儿第3、第4次美罗华治疗。移植后+59天,PETCT 报告示右侧颈部多发淋巴结肿大伴氟代脱氧葡萄糖(FDG)代谢增高,考虑淋巴瘤累及;双侧颌下及左侧颈深淋巴结FDG代谢未见明显增高;全身其他部位除散在出血及炎症渗出病灶外未见异常(见图2)。在积极给予患儿抗病毒+减抗排异药+利妥昔单抗抢先治疗(4疗程)后,其外周血EBV-DNA已转阴,但颈部的肿大淋巴结并无明显缩小,考虑患儿EBV-PTLD治疗后部分缓解(PR)。遂给予低剂量化疗方案。移植后+70天起,予患儿第1疗程低剂量化疗:+70天,环磷酰胺(CTX)600 mg/m2;+71天,利妥昔单抗375 mg/m2;+71天~ +75天,甲基泼尼松龙0.8 mg/(kg·次),q12h。移植后+69天、+73天、+80天、+87天,患儿外周血EBV-DNA 仍均为阴性;移植后+86 天,患儿颈部增强CT检查报告示双侧颈部仍可见散在肿大淋巴结,较大者16.3 mm × 10.1 mm。移植后+96天,予患儿第2疗程低剂量化疗,过程顺利。移植后+111天,患儿颈部增强CT检查报告示双侧颈部未见肿大淋巴结。移植后+121天,予患儿第3疗程低剂量化疗(CTX+甲基泼尼松龙),过程顺利。此后,分别间隔21 天,患儿顺利完成第4~6疗程化疗。
目前,患儿EBV-PTLD经治疗后已完全缓解(CR)6月,定期门诊随访,一般情况可,最近一次复查血心肌酶、肝肾功能、电解质、血常规等指标均未见明显异常。
2 讨论
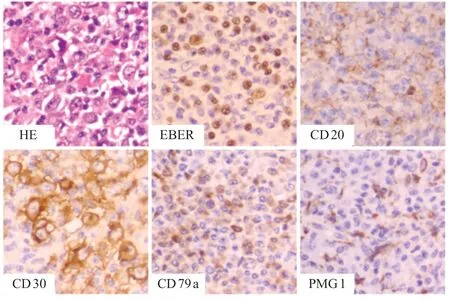
图1 患儿右侧颈部肿大淋巴结病理活检组织化学染色(×400)
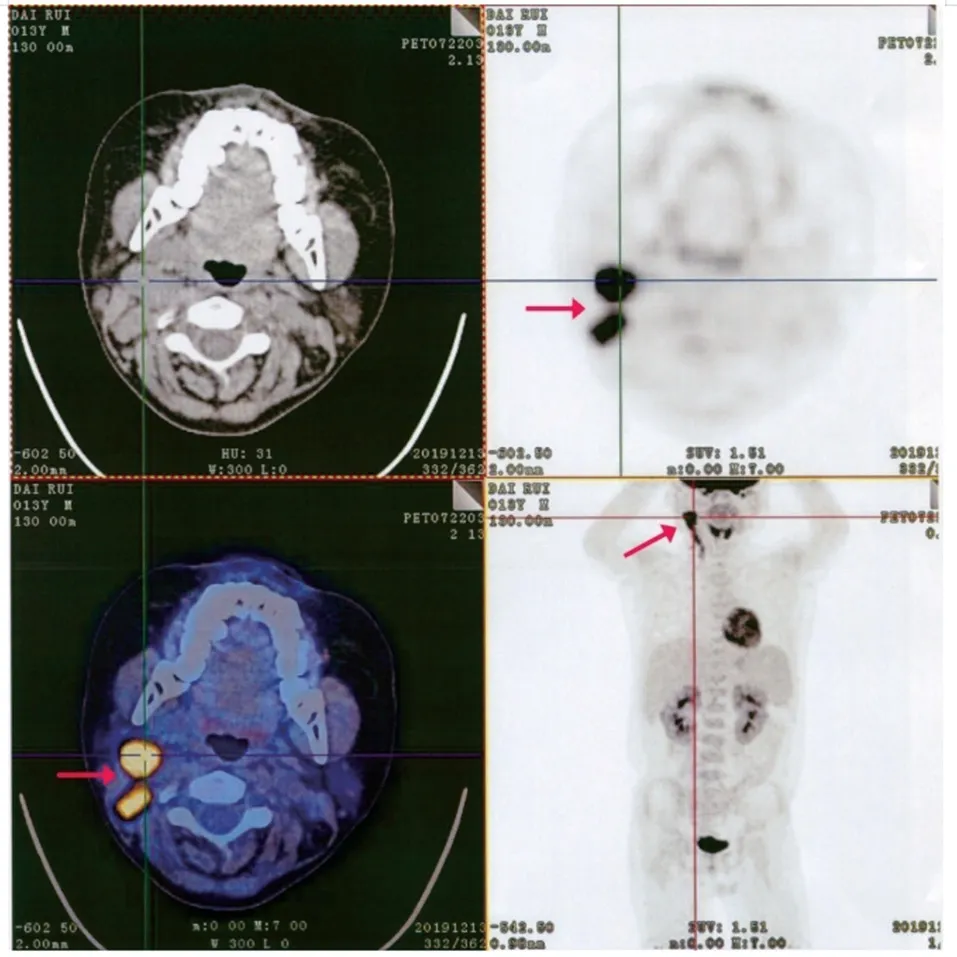
图2 患儿全身PET-CT 影像学表现
EBV属于γ-疱疹病毒,广泛分布于自然界中,易感染人类B 淋巴细胞,甚至可以长期潜伏[2]。EBV 可以通过唾沫传播。在我国婴幼儿中,EBV-IgG抗体阳性率达90%以上。免疫力正常的患儿,可以长期没有临床表现或者症状很轻,但是HSCT后患儿免疫功能低下,容易出现EBV再活化及感染,甚至可能进展为致命性的PTLD。目前认为PTLD 的发生是由于移植后EBV 特异性杀伤T 细胞功能受损或免疫重建延迟导致[3]。
流行病学研究发现,近十年儿童PTLD 的总体发病率为4%左右,但有高危因素患儿的PTLD 发病率在此基础上提高10%~20%。目前已被证实的PTLD高危因素[3]包括:①低强度预处理方案。因为低强度预处理的患儿HSCT后需要增强抗排异治疗,容易导致EBV 特异性细胞毒性T 细胞重建延迟,从而诱发EBV血症甚至PTLD。②T细胞去除(包括阿仑单抗、梯度/密度离心等)。T 细胞去除在移植预处理和预防GVHD 方面被广泛运用,但是移植前使用和未使用T细胞去除,患儿移植后其PTLD发病率分别为≥10%、<1%[3]。③使用ATG 的移植患儿中有1.7%会发生PTLD[3],是一个独立危险因素。④单倍体半相合供者、≥2个HLA抗原错配及无关供者,受者PTLD发病率分别为(1.3±0.7)%,(13.7±1.7)%和(9.1±4.4)%[3]。⑤Ⅱ~Ⅳ级GVHD。中重度GVHD 会引发炎症风暴,削弱患儿特异性免疫功能,从而导致PTLD[4]。⑥供受者血清学EBV 错配。例如,给EBV 血清学阴性的移植受者输注来源于EBV 血清学阳性的移植供者的造血干细胞后,患儿发生PTLD 的风险明显增高[5]。⑦全身放疗也是PTLD的独立危险因素[6]。⑧脐血移植。目前脐血移植的预处理方案通常是低强度预处理方案,且包含ATG 的使用,故其为PTLD 发生的高危因素。⑨感染的EBV 载量。移植后EBV 载量较高或者较低的患儿总生存期比EBV 中度载量的患儿要明显延长[7]。⑩CMV感染。移植后CMV阳性的患儿有32.3%会出现EBV激活,而移植后CMV阴性的患儿仅有19%会出现EBV激活[8]。○11 脾切除。脾切除削弱了CD 5+B 细胞的免疫功能,可导致EBV 的爆发性增殖[2],从而进一步发展为PTLD。○12 原发疾病的影响。例如,重型再障(SAA)患儿移植前有ATG 和环孢素治疗史,因此其PTLD 发病率较高[3];非霍奇金淋巴瘤患儿移植前有利妥昔单抗治疗史,其PTLD 发病率反而偏低[9]。
WAS是一种罕见的X-连锁隐性遗传免疫缺陷病,allo-HSCT是目前唯一的治愈方法。免疫缺陷病HSCT治疗目标是:在供体造血干细胞能够完全植入的前提下,尽量减少移植后的免疫排斥反应,改善患儿的生活质量。因此,本例患儿选择清髓性的预处理方案(包含ATG)和骨髓库HLA配型全相合的无关供者。本例患儿预处理方案中ATG的使用、来源于骨髓库无关供者的造血干细胞以及移植后的GVHD 可能导致患儿EBV特异性细胞毒性T淋巴细胞免疫重建延迟,最终诱发EBV 血症及PTLD。PTLD 的常见临床表现包括持续发热、浅表淋巴结无痛性肿大、扁桃体肿大、间断性腹痛、频发呕吐、腹泻、肝脾肿大、抽搐等症状,部分患儿可仅表现为淋巴结肿大,而无其他明显症状。实验室检查常见EBV-DNA 拷贝数连续2 次>103拷贝/mL、血常规三系或两系减少、肝肾功能异常、乳酸脱氢酶增高等异常。根据2016年世界卫生组织(WTO)新分类[10],EBV相关的PTLD病理上可以分为4种基本类型:①早期非破坏性PTLD,包括反应性浆细胞增生、传染性单核细胞增多症样的PTLD 及明显滤泡增生;②多形性破坏性PTLD,多数细胞EBER检测阳性,分子检测可见Ig基因重排,但形态学还没有达到淋巴瘤的诊断标准;③单形性破坏性PTLD,最常见,包括弥漫大 B 细胞淋巴瘤、Burkitt 淋巴瘤、浆细胞瘤,已达到诊断淋巴瘤标准;④经典霍奇金淋巴瘤型 PTLD,罕见,其组织形态学已满足经典型霍奇金淋巴瘤的诊断标准,病理切片中可见镜影(Reed Sternberg)细胞。
目前针对儿童EBV-PTLD 的治疗主要包括两个方面:一方面是对可疑PTLD 的早期治疗,即有≥2个高危因素的预防性治疗和针对EBV 血症的治疗;另一方面是组织病理活检确诊后的治疗。现行的治疗手段[11]包括:①减抗排异药(RIS)。有效率最高可达73%,但其中位起效时间大约3~5 周[2],而对于预处理前接受过T 细胞去除处理、ATG 或者阿伦单抗治疗的患儿治疗效果欠佳。②利妥昔单抗是抗人CD 20 的单克隆抗体。预防性应用利妥昔单抗可明显降低PTLD 的发病率。预防措施包括T 细胞去除 处理的受者移植前给予预防性低剂量利妥昔单抗治疗(100 mg/m2)[12];在外周血EBV-DNA阳性前或者第1次出现外周血EBV-DNA≥1 000 拷贝/mL时,即给予足剂量利妥昔单抗治疗(每周375 mg/m2,1~4 周)[13]。③细胞免疫治疗。如EBV 特异性细胞毒性T细胞输注疗法(CTLs),有高危因素的移植受者预防性使用CTLs成功率达100%;而对已经确诊PTLD的患儿,治疗后CR率也达84.6%,尤其适用于PTLD复发或者对利妥昔单抗耐药的患儿,均疗效显著[14]。新一代的CTLs,被引入“自杀基因”后,不会诱发因输注EBV-CTLs 而导致的GVHD[2,14]。嵌合抗原受体T 细胞(chimeric antigen receptor T cells,CAR-T)免疫治疗,即通过提取改造患者自身免疫系统的T细胞,使其表达嵌合抗原受体(CAR),来识别特异性的肿瘤细胞靶点,并释放大量γ-干扰素激活T细胞功能,最终实现肿瘤杀伤。最新研究证实抗CD19 CAR-T治疗复发难治性大B细胞淋巴瘤的总有效率约80%,完全有效率约50%,治疗后1~2年无病生存率为35%~40%[15-17]。供体淋巴细胞输注(donor lymphocyte infusion,DLI),可以有效治疗EBV-PTLD,但是EBV-PTLD患儿接受DLI 治疗后,GVHD 发生率高达17%[2],因此临床诊疗过程中须密切关注DLI引发严重GVHD的风险。④化疗,包括低剂量化疗和传统化疗,常用于患儿PTLD复发或者对利妥昔单抗疗效欠佳的抢救治疗。但是目前国内还没有形成统一的化疗方案,正处于不断地探索改进中。⑤抗病毒治疗(包括更昔洛韦、缬更昔洛韦、膦甲酸钠氯化钠等抗病毒药物的使用)。这些抗病毒药物的主要作用机制是抑制病毒复制,诱导细胞 凋亡[18]。
综上所述,EBV-PTLD是儿童HSCT后的一种严重并发症,进展迅速,死亡率高,预后也差。本例WAS患儿HSCT 后出现外周血EBV 阳性时,通过积极给予抗病毒治疗,逐渐减量抗排异药,以及利妥昔单抗和低剂量化疗的联合应用,取得了比较满意的治疗效果。由于儿童EBV-PTLD发病率低、相关临床试验少等原因,目前国内还没有形成明确的诊疗共识,仍然需要更多前瞻性研究来完善儿童EBV-PTLD 治疗的有效性和安全性。
猜你喜欢
传染病信息(2022年3期)2022-07-15
新医学(2022年4期)2022-04-23
中国典型病例大全(2022年9期)2022-04-19
现代临床医学(2022年2期)2022-04-19
健康体检与管理(2022年2期)2022-04-15
昆明医科大学学报(2022年2期)2022-03-29
中国药学药品知识仓库(2022年2期)2022-03-23
现代临床医学(2021年4期)2021-07-31
感染、炎症、修复(2021年1期)2021-07-28
康颐(2020年1期)2020-09-10
